The 12th AD/PD conference drew some 3,100 people from across the world to the Mediterranean city of Nice. After a long day of talks—sessions ran from 8:30 a.m. to a grueling 7:15 p.m.—hungry scientists could be seen strolling to dinner across the Place Massena, with its illuminated resin statues created by the Catalan contemporary artist Jaume Plensa. The seven characters on their pedestals represent seven continents, and the changing color of their lighting is said to symbolize communication between them. At the conference, talks in four parallel sessions ran the gamut from basic to clinical science on Alzheimer's, Parkinson's, the in-between disease dementia with Lewy bodies, and even a session on the ALS-FTD spectrum.
Biogen Antibody Buoyed by Phase 1 Data and Hungry Investors
Last week in Nice, France, a single story took the 12th International Conference on Alzheimer’s and Parkinsons’s Diseases by storm. Everyone who was not on Mars on March 20 probably heard from their local radio or TV station, Twitter, or favorite online source that BIIB037, aka aducanumab, the anti-Aβ antibody developed by the company Biogen in Cambridge, Massachusetts, improved Alzheimer’s symptoms in a large Phase 1 trial. Expectations had been high ever since a Biogen executive told investors at a bank conference in Boston last December that aducanumab was showing a dose-dependent benefit on both amyloid removal and cognition.
Amyloid PET scan in a BIIB037 trial participant at baseline (left) and after one year on treatment (right). [Image courtesy of Biogen.]
That tease was enough to jack up Biogen’s stock and add billions to its market capitalization prior to the AD/PD conference, which was held March 18 to 22. In Nice on the two days before the aducanumab presentation, prominent Alzheimer’s researchers bantered about how financial analysts were offering them hourly rates up to four figures for comments immediately after the talk. Some analysts were flying in from New York City for the day. During the presentation by Biogen’s Jeffrey Sevigny, one could hear a pin drop in a full auditorium that seats 2,500. The same morning, stories went live online, and the blogosphere and Twitter exploded with chatter. Biogen’s stock price jumped by 7 percent in online trading between Sevigny’s talk at 5:25 a.m. NYC time and the opening bell on Wall Street. It temporarily rose even further before settling there once a heady mix of real results and hyperbole had run its course. In the Nice convention center, some researchers poked fun at the spectacle, waving their arms and shouting: “Sell now! Sell now!” In earnest, however, most scientists were electrified by half of the data Sevigny presented and appropriately cautious about the rest. So what’s the real story? Here is a summary of the data, and what leading researchers think about them.
Formerly called BIIB037, aducanumab is a high-affinity antibody against Aβ. Like its unfortunate cousin bapineuzumab, aducanumab recognizes Aβ’s N-terminal amino acids 3 to 6; however, its slightly wider binding pocket recognizes a structural epitope that is present on aggregated Aβ but absent from monomers. Biogen licensed BIIB037, along with antibodies against aggregated tau and α-synuclein, from the Swiss biotech Neurimmune, picking off some of Neurimmune’s larger panel of antibodies derived from healthy aging donors (for more on the approach, see Apr 2013 conference news).
Aducanumab is notable for its long half-life in human plasma without getting stuck to monomeric plasma Aβ, Neurimmune’s founder Roger Nitsch said at AD/PD. This is important, he said, because it allows aducanumab to accumulate in the brain and find its target there despite antibodies’ generally poor ability to cross the blood-brain barrier.
At AD/PD, Sevigny presented an interim analysis of some of the data of a Phase 1b trial. This was not a final analysis, but merely the first of several data cuts pre-specified in the protocol for this ongoing study, Sevigny emphasized to Alzforum. Clinicaltrials.gov shows this study as collecting data until May 2016.
Following a previous single-ascending-dose study, this larger Phase 1b trial enrolled 166 people whose early Alzheimer’s diagnosis was confirmed by way of a positive scan with the amyloid PET tracer florbetapir. This trial followed a staggered parallel group design, where increasing dose cohorts follow each other. In cohort 1, people were randomized to 1 mg/kg or 3 mg/kg of antibody, each 3:1 to placebo; then an additional cohort was randomized to 10 mg/kg or placebo. Partway into the trial, Biogen followed a recommendation of its data safety-monitoring board and added a third cohort of 6 mg/kg because the highest dose was showing more amyloid-related imaging abnormality, or ARIA-E, a closely watched side effect of anti-amyloid therapy.
This latest cohort has reached its pre-specified first data cut, taken at 30 weeks, but not yet the next cut, which will come at 54 weeks, Sevigny said. Therefore the six-month outcome data he presented at AD/PD compared all doses, but the one-year data did not yet contain the 6 mg/kg cohort. In addition, the trial added a titration cohort in search of ways to minimize the risk of ARIA-E. The placebo-control period of the study involves one injection every four weeks, for a total of 14; participants who complete the one-year blinded portion can choose to add a year of dose-blind extension.
For outcomes, the study set safety and tolerability as the primary, and six-month change in amyloid PET as the secondary endpoints. Exploratory endpoints included this formidable list: one-year amyloid PET; MMSE; CDR-sum of boxes; a version of the NTB neuropsych battery; the FCSRT (which doubled as an inclusion criterion); the NPI-Q neuropsychiatric inventory; and small sub-studies on fluid biomarkers, FDG PET, as well as volumetric, resting-state functional, and ASL spin labeling MRI. On top of that came a quarterly surveillance MRI to monitor ARIA-E.
The groups were well-balanced across arms for baseline characteristics, Sevigny said, with a mean age in the low 70s, a mean MMSE of 25, and mean PET uptake of 1.44 as expressed by composite SUVR. Clinically, about 60 percent were diagnosed as having mild AD; two-thirds carried at least one ApoE4 allele.
At AD/PD, Sevigny showed that amyloid deposition nudged up slightly in the placebo group, but dropped in all treatment groups at week 26 and even more at one year. At six months, this was statistically significant for the 3, 6, 10 mg/kg arms, and at 12 months for the 3 and 10 mg/kg arms. Amyloid was cleared in each of the six cortical regions of interest the scientists measured—frontal, parietal, lateral temporal, sensorimotor, anterior, and posterior cingulate. Importantly, the magnitude of this reduction was far greater than the dips in amyloid load reported in previous trials of bapineuzumab (Rinne et al., 2010) and gantenerumab (Ostrowitzki et al., 2012). In fact, after one year, the 10 mg dose appears to have pulled most amyloid from the brain as that group’s SUVR neared a commonly used threshold of positivity, set here at 1.13. When plotted as amyloid load over time, the groups spread apart by dose, albeit without the results of the 6 mg/kg dose at one year. “Target engagement has been demonstrated,” said Sevigny.
Next, Sevigny showed data for the MMSE and the CDR-SB. The former is a quick screen that taps different domains of cognitive function, the latter is a mixed cognitive/clinical instrument that includes some informant input. On the MMSE, the arms stayed closely together at six months, but by one year they had separated. The placebo group had worsened by 3.1 points, the 1 mg/kg group by about 2 points and the 3 and 10 mg/kg doses by less than 1 point. The two higher-dose groups appeared to stabilize after six months. On the CDR-SB, too, the groups were still together at six months, but by one year they had separated in a dose-dependent way, again with the 6 mg/kg result still pending. On this measure, the placebo group worsened by 2 points and the highest dose by about 0.5 points. The highest dose result was statistically significant and appeared to indicate stabilization. “This supports our hypothesis that amyloid reduction confers a clinical benefit,” Sevigny said.
On safety, Sevigny reported that three patients died, of metastatic cancer, heart attack, and complications from hip surgery, respectively. Two had been on placebo, one in the high-dose group. The study physicians considered the deaths unrelated to aducanumab, Sevigny said. The most common side effect was ARIA-E, a finding of white spots in the magnetic resonance image that reflect vasogenic edema, e.g., fluid in the parenchyma. The surveillance MRIs showed lots of those, mostly in the ApoE4 carriers and at higher doses: one in the 1 mg/kg group, two in the 3 mg/kg group, 10 in the 6 mg/kg group and 13 in the 10 mg/kg group. This was a surprise after the first, smaller trial (see Apr 2013 conference news). Most ARIA-E occurred within the first five doses. Two-thirds were asymptomatic, but one-third caused mild to moderate headaches, visual disturbances, or confusion, which usually resolved within a month, Sevigny said.
“Most participants were unaware that they had ARIA-E,” Sevigny told Alzforum. That is because they did not come in to be seen for symptoms of ARIA-E; rather, their next scheduled MRI picked it up and clinicians then asked if the patients had experienced specific symptoms in the past month. The trial’s protocol guided the study physicians to let patients continue at the same dose, at a lower dose, or to stop dosing, depending on how severe the symptoms were. About half of the participants who developed ARIA-E continued treatment and had no additional ARIA-E; however, one person in the 1 mg/kg group, three in the 6 mg/kg group, and eight in the 10 mg/kg group discontinued treatment. Most were ApoE4 carriers, Sevigny said.
There was little public discussion after Sevigny’s talk, but as soon as he stepped off the lectern, conversation bubbled up and continued for days in the hallways, on the escalators, and over meals. Scientists talked of little else. Initial responses were enthusiastic and became a bit more tempered as time allowed the limitations of this 20-minute presentation to sink in. The Karolinska Institute’s Bengt Winblad from Stockholm spoke for many a battle-weary site leader when he said, “These data for the first time in a long time seem to be positive.” Dennis Selkoe of Brigham and Women’s Hospital in Boston exclaimed, “I am happy! To me a key point is that the amyloid went down at six and even more at 12 months, and that from six months on cognitive decline appears to have been arrested at the highest dose.” Colin Masters of the University of Melbourne in Australia called the data “a game-changer,” and Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center in Las Vegas noted, “This is the first time we have seen both MMSE and CDR-SB appear to go in the same direction, strongly supported by a biomarker.”
John Hardy at University College London opined, “It was great to see a convincing dose response in every measure. This must give hope to other companies that if you reduce amyloid enough, there will be a clinical benefit.” Enchi Liu of Janssen Research and Development, LLC, agreed. “This is good news for us all. It validates the class of drugs,” she said. Liu worked on bapineuzumab, whose dosing was limited by concerns over ARIA-E; it showed modest target engagement but no efficacy in Phase 3. Many other researchers at AD/PD were similarly relieved at finally getting a shot in the arm after years of setbacks. “This looks good, so long as we remember it’s early days,” said Fred Van Leuven, who co-founded AC Immune, the company that originally developed the Aβ antibody crenezumab and several tau-based immunotherapies. “We are receiving inquiries from people who want to join trials,” said Creighton (Tony) Phelps of the National Institute on Aging in Bethesda, Maryland.
The market’s ebullience will help Biogen finance expensive Phase 3 trials. That said, more than 20 scientists this reporter spoke to were at pains to avoid irrational exuberance. “We have been burned before,” was a common refrain. Most sources drew a dividing line between the strength of the biomarker and safety data, on the one hand, and the limitations of the clinical/cognitive data disclosed thus far.
The target engagement data was widely praised. A large, dose-dependent, and consistent reduction of brain amyloid is a huge step forward, scientists agreed. “They pretty much cleared amyloid out of the brain. That is fantastic, provided it can be repeated,” said Chris Rowe, a PET expert at the University of Melbourne. Besides the SUVR mean value and range for each group, Rowe would like to see the SUVR trajectories of individual participants rendered as spaghetti plots, the way ADNI and AIBL show their data. Amyloid PET data in past cohort studies and clinical trials were marked by considerable variability, particularly when using the brain’s cerebellum as a reference region (see Dec 2014 conference news). Spaghetti graphs show both variation and overall direction at a glance. Also, by depicting each patient’s time course, spaghetti graphs can prove that a few patients did not drive the group result, particularly in the face of a significant number of treatment discontinuations. Other PET experts said the florbetapir data looked cleaner than anything they had ever seen, probably because the drug works so well, but almost to the point where one might begin to worry about a hidden systemic bias. Some scientists wondered if the antibody might bind its epitope so strongly that it edges out florbetapir binding, but Biogen researchers replied they had ruled that out in a competition binding study with Avid Radiopharmaceuticals, the maker of the tracer, before starting the clinical trial.
Of the safety data, the trial’s rate of ARIA-E generated the most debate. Some clinicians thought this side effect was so common at the higher doses that regulatory agencies in some countries might balk at approving a Phase 3 in 2015, insisting on more dose finding or waiting out a first Phase 3 in the United States. Others noted that the field had come a long way in the past decade since ARIA cropped up during the development of bapineuzumab. The key advance now with aducanumab is its strong evidence of target engagement and a clinical signal. Given that, many believe the conversation will evolve from “ARIA is an unacceptable risk” to “How do we best manage ARIA?”
At AD/PD, Janssen’s Liu reinforced this notion. She analyzed bapineuzumab’s Phase 3 dataset to learn if any baseline markers predicted who would develop ARIA-E. No marker did, Liu reported in her talk, but she also found that people who developed ARIA-E subsequently had larger changes in their downstream biomarkers of neurodegeneration, such as CSF tau and brain volume, than those who did not. This strengthens the view that ARIA-E indicates that the antibody is biologically active. The challenge then becomes one of finessing the dose and timing of the injections in such a way that the patient suffers no harm but still gets the benefit. In discussion, some scientists asked if the target engagement and apparent efficacy of aducanumab will spur a change in the paradigm of AD therapy toward the way cancer therapy is viewed. There, the doctor tells the patient: “I will make you very sick before I make you better,” and regulators and society at large accept that.
In all, scientists considered the primary and secondary endpoints of this trial a success. “This trial provides important information about safety and target engagement. This is great medicinal chemistry. They have a dose response and a handle on dosing for the next trial. What more can you ask from a Phase 1b?” said Lon Schneider of the University of Southern California.
A lot more, it appears. At AD/PD, Biogen researchers clearly stated that the trial was not powered for cognitive or clinical outcomes. In fact, Biogen’s trialists had initially designed the study to assess only safety and target engagement, as is customary in Phase 1, but were directed to add exploratory clinical measures. Those are what the investment market and the media latched onto, treating this Phase 1b study as a de facto Phase 2. And Biogen’s leaders in effect did so, too, by deciding to forgo a Phase 2 trial and go straight to Phase 3.
At AD/PD, many scientists pointed out that the study was small. Cognitive outcomes are noisy, and carry little information at fewer than 40 patients per group, said Schneider. As per protocol, Biogen handled the data from these measures by adjusting every person’s actual MMSE and CDR-SB baseline values to a point 0 and scoring change from there together with a p value, rather than plotting the actual values. Some commentators would have preferred to see more raw data of participants’ actual MMSE and CDR-SB values, but others considered this a routine way of presenting such datasets. All agreed the cognitive data was but a preliminary signal.
Others bemoaned that other exploratory outcomes were not presented at AD/PD. When pressed about that, several Biogen scientists assured Alzforum that they simply have not had time yet to analyze those data.
Some pharma scientists considered the decline of the placebo group surprisingly large, given that many patients in the mildest symptomatic stages barely change in a year, especially if they take concomitant medications. However, others disagreed, saying that those expectations come from more heterogeneous cohorts where a proportion has no underlying Alzheimer’s pathology, whereas all participants in this trial did (e.g. Coley et al., 2011).
Some scientists would have liked to see ADAS-cog data to compare aducanumab directly to the crenezumab ABBY trial, which was negative overall but found an efficacy signal in the mild AD subgroup (see Jul 2014 conference news). About half of the patients in the aducanumab trial are at roughly the same mild AD stage. The aducanumab trial did not use the ADAS-cog. In 2013, the Food and Drug Administration issued a guidance for drug development in earlier stages of AD (see Mar 2013 news) that stated that trialists could use a single primary outcome such as the CDR-SB, which includes both cognitive and functional measures. Biogen ran with it, and provided the first evidence that the CDR-SB is sensitive in a treatment study at this stage.
For all outcomes—amyloid removal, cognition and function—scientists puzzled over the treatment discontinuations in the highest dose. Missing data is a common problem in randomized controlled trials. When a disproportionate fraction of a particular group drops out, how the analysis handles that becomes important for the overall result of the trial. The suspicion is that if the worst responders drop out from a treatment but not placebo arm—in this case, for example, the ApoE4 carriers with symptomatic ARIA-E from the high dose—it could make the drug look better than it is. Common ways of analyzing such datasets include:
Intent-to-treat, aka ITT—data from every randomized person goes into the analysis regardless of whether they received study drug;
Last observation carried forward, aka LOCF—missing data are imputed based on the last available data from any given patient;
Per-protocol analysis, where only the data from people who got all treatments and appeared for all visits is included.
Sevigny’s slides, which were not made public but nonetheless circulated as people snapped pictures on their tablet computers, stated in fine print at the bottom that the analysis was based on observed data. Sevigny explained to Alzforum that the aducanumab trial used a modified ITT based strictly on data in hand. That is, it included data from everyone who was randomized, received at least one injection, and underwent at least one set of subsequent assessments, regardless of how many injections they received or whether they completed all visits. People who discontinued treatment were urged to come for their scheduled assessments anyway, and those data went into the analysis. In other words, treatment discontinuation is not the same as dropout. When someone skipped a visit or dropped out for good, missing data were not imputed, Sevigny said. So hypothetically, the data from a person who received four injections, developed symptomatic ARIA-E, discontinued treatment, but came for his or her 54-week assessment, would all go into the analysis. Likewise, the data from a participant who failed to show up for his or her 54-week visit would simply be missing from the pool, rather than estimated based on his or her prior trajectory.
All in all, the scientists were immensely pleased about the results for which the study was powered, and cautious about the rest. “Let’s not overvalue the clinical outcomes. Biogen was careful to say they are exploratory. The rest of the world seems to think we have to make inferences on the clinical outcomes as they were presented. We will be able to assess the clinical outcomes soon enough in the Phase 3 trials,” said Schneider. By March 24, analysts on Twitter had calculated aducanumab’s potential worth at $90 billion.—Gabrielle Strobel
Antibody Against α-Synuclein Looks Safe In Phase 1
People with Parkinson’s disease have treatment options, but none directly tackle the underlying pathology of misfolded and aggregated α-synuclein. That may be poised to change, with numerous approaches targeting the protein now in the pipeline, including two immunotherapies in the clinic. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18–22 in Nice, France, researchers at Prothena Biosciences of South San Francisco, California, presented topline Phase 1 data from their anti-α-synuclein antibody. It’s early days, but at first glance, at least, the treatment appears safe in healthy volunteers and squelches serum levels of the protein. Researchers will have to wait for the results of an ongoing trial in Parkinson’s patients to learn how the treatment behaves in the central nervous system. Meanwhile, an active vaccine under investigation by the Austrian company AFFiRiS, Vienna, has cleared Phase 1.
Published in 1892, this photograph shows the characteristic posture of a patient with Parkinson’s disease.
Other speakers described different preclinical strategies to normalize α-synuclein. Overall, the AD/PD talks showcased a field that has placed α-synuclein squarely in its sights even while some crucial questions remain unanswered. Chief among these is determining exactly what its toxic form is. “It’s exciting to see clinical progress made toward α-synuclein-lowering biologics,” Clemens Scherzer at Harvard Medical School wrote to Alzforum.
Why the emphasis on targeting α-synuclein now? In short, because researchers finally can. Alpha-synuclein has drawn intense interest ever since the late 1990s, when the first α-synuclein missense mutation was identified as a cause of familial Parkinson’s and the protein soon after turned out to be the main constituent of Lewy bodies. But α-synuclein has been an obstreperous target because it has been slow to give up its secrets about what it does normally and in disease. Finally, this year, some basic knowledge and tools are in hand, and companies developing immunotherapies against α-synuclein include, besides Prothena and AFFiRiS, AstraZeneca, BioArctic, Biogen, Genentech, and Lundbeck.
In animals, researchers know that suppressing toxic forms of the protein reverses cognitive and motor deficits (Jul 2011 news; Nuber et al., 2013). Also in mice, antibodies can stop the transfer of misfolded α-synuclein from cell to cell, improving neuron survival as well as motor function (Jun 2014 news). A therapy against pathological α-synuclein would benefit not only PD patients, but also people who have dementia with Lewy bodies (DLB) or multiple system atrophy (MSA), and perhaps even some Alzheimer’s patients, noted Eliezer Masliah of the University of California, San Diego.
Immunotherapy approaches are currently closest to the clinic. At AD/PD, Wagner Zago, who leads Prothena’s research division, said his team has been working on an antibody for human use since 2003. Prothena is an outgrowth of Athena Neurosciences and Élan Corporation. Some of its employees, notably Dale Schenk, originally developed immunotherapy for Alzheimer’s disease, such as the first active vaccine, AN-1792, and the antibody bapineuzumab.
In preclinical work, the researchers found that targeting different epitopes of α-synuclein gave varying efficacy. “Most of the antibodies we tested failed,” Zago noted. Those that recognized the C-terminus of α-synuclein performed best. Cell culture experiments suggested that these antibodies somehow prevented the protease calpain-1 from cleaving the C-terminal end of extracellular α-synuclein, Zago said. Accumulation of cleaved forms in particular correlates with aggregation and toxicity (Mishizen-Eberz et al., 2003; Li et al., 2005; Dufty et al., 2007).
A Lewy body and neurite from Parkinson's brain containing α-synuclein (green=full length; red=cleaved) and proximal to nuclei (purple). [Image courtesy of Prothena Biosciences Inc.]
The winning antibody from preclinical studies was 9E4, and its humanized version became PRX002. It is an IgG1 monoclonal that binds better to aggregated forms than to monomers, Zago reported. In mice, it lowered α-synuclein deposits and gliosis, while improving spatial memory and motor skills (Games et al., 2014).
The findings led Prothena to start Phase 1 trials in collaboration with Hoffmann-La Roche in Basel, Switzerland. At AD/PD, Zago presented data from a single-ascending-dose study in 40 healthy volunteers. The study tested intravenously administered doses of 0.3, 1, 3, 10, and 30 mg/kg in six participants each, with the remaining 10 volunteers receiving placebo. Most importantly, Zago said, this first experiment with an anti-α-synuclein antibody was safe and well-tolerated at all doses tested. The antibody had favorable pharmacokinetic properties. It appeared to engage its target in the periphery, because it caused free α-synuclein in the blood to plummet by as much as 96 percent, Zago said. The reductions were dose-dependent, he said in answer to an audience query, though he stayed mum on details. A March 19 press release from Prothena noted that the main adverse events were headache and pain at the injection site.
Some audience members worried whether antibodies interfered with the cellular and synaptic functions of normal α-synuclein in the brain. Zago answered that Prothena’s preclinical studies found no change in the levels of monomeric α-synuclein levels in the brain during antibody therapy. In a panel discussion, Masliah, who has collaborated with Prothena on preclinical studies, noted that α-synuclein participates in synaptic fusion and neurotransmitter release. These functions continued normally in cell cultures after treatment with PRX002, he said.
Scherzer wrote to Alzforum that this trial does not yet answer the key question. “The reported reduction of free serum α-synuclein levels … is really intriguing and shows that there is a peripheral pharmacodynamic effect. However, α-synuclein is selectively and highly expressed in red blood cells … We don’t know yet whether the target was engaged centrally,” he wrote. The answer should be forthcoming. PRX002 is currently in a Phase 1 multiple-ascending-dose study in 60 Parkinson’s patients. That study includes cerebrospinal fluid (CSF) collection. “In this first trial we were mostly concerned to show that the antibody engages α-synuclein and that that is a safe thing to do. Everything else we still need to show,” Zago told Alzforum.
AFFiRiS’ active vaccine PD01A recently completed Phase 1 testing of multiple doses in 32 Parkinson’s patients (Mandler et al., 2014). A poster at AD/PD reported the treatment as being safe and producing antibodies in 15 of the 24 participants who received the vaccine. According to a July 2014 press release, a follow-up Phase 1 study is still ongoing. A second poster noted that AFFiRiS is also testing the vaccine in a Phase 1 trial of MSA patients. In this disease, α-synuclein accumulates in oligodendrocytes, leading to demyelination and movement problems. In a mouse model, treatment with PD01A mopped up α-synuclein aggregates in oligodendrocytes, activated microglia, and preserved myelination and neuron health, the scientists reported.
In Nice, Masliah outlined other ways to go after α-synuclein. They include blocking aggregation, promoting degradation, stabilizing non-toxic forms, or preventing toxicity by targeting receptors. “Different therapies will be applicable to different stages of the disease,” Masliah predicted.
Blocking aggregation has been popular with researchers, and several such inhibitors are inching their way toward the clinic. The Seattle-area company ProteoTech plans to take its aggregation inhibitor Synuclere, which is administered subcutaneously, into Phase 1 trials for MSA. The compound busts up α-synuclein deposits and improves motor skills in old Parkinson’s model mice, ProteoTech’s Alan Snow reported at AD/PD (Apr 2011 conference news). Snow told Alzforum that he is seeking collaborations with pharma companies. He is working on an oral formulation of the compound before moving into PD trials.
Armin Giese at Ludwig-Maximilians Universität, Munich, and Christian Griesinger at the Max Planck Institute in Göttingen formed the company MODAG GmbH in Wendelsheim, Germany, to develop the general aggregation inhibitor Anle138b. They plan to start clinical trials within the year (Apr 2011 conference news; Aug 2014 conference news). At AD/PD, Michal Wegrzynowicz of Cambridge University in England presented new data on the compound’s efficacy in a mouse that expresses truncated human α-synuclein. Dopamine levels drop in these animals at six months of age, and by 12 months, dopaminergic neurons are dying. In contrast, animals that ate Anle138b in their chow from 9 to 12 months of age maintained their neurons, and levels of the neurotransmitter rebounded, Wegrzynowicz reported. The result complements recent work from Giese and Griesinger, which found that treatment with Anle138b prolonged survival of year-old, symptomatic mice that model PD by about 2 months (Levin et al., 2014).
Even though some α-synuclein therapeutics are in, or within reach of, clinical trials, researchers acknowledge a pressing need for better biomarkers to gauge their efficacy. Gene Kinney, who spun out with Prothena from Élan, told Alzforum that, as in the AD field, early trials will include a plethora of candidate biomarkers to find those that track most closely with clinical improvement. At AD/PD, Mark Frasier of the Michael J. Fox Foundation discussed the organization’s efforts to help in this regard. The MJFF has invested $100 million in biomarker discovery and validation, Frasier noted (Oct 2010 news series; Mar 2014 conference news). Foundation projects are attempting to standardize research-grade serum and CSF assays of α-synuclein, improve their sensitivity and dynamic range, and move promising “home brews” toward mass production, Frasier said. One such assay, developed by Michael Schlossmacher and Brit Mollenhauer and then mass-produced by BioLegend, is commercially available and being used in the Parkinson’s Progression Markers Initiative; Prothena is developing in-house assays with Roche to use with PRX002.
Kinney noted that trial results in Alzheimer’s have shown the importance of screening participants for trials to make sure they have the pathology being targeted by the investigational drug. Screening is done with CSF or amyloid PET. A research consortium funded by MJFF to develop PET tracers for detecting the protein in living brains (Bagchi et al., 2013) remains a ways off from human trials. One challenge in finding a high-affinity tracer—and even just sufficiently robust and specific antibodies—is that they tend to bind other amyloids in brain tissue, which in many α-synucleinopathies comprise a mixture of misfolded protein deposits. “For Parkinson’s, we need even better biomarkers than for Alzheimer’s, because the mixed pathologies are a bigger problem in PD than AD. People enter the clinic with movement disorders and not everything is due to α-synuclein,” said Lars Lannfelt of Uppsala University, who works on a similar antibody to Prothena’s with the company BioArctic.
Not everything is more complex in Parkinson’s, however. One consolation is that α-synuclein does not form extracellular deposits on blood vessels, so sides effects such as ARIA are unlikely to be a concern.
One burning issue flared up in many talks: The pathological species of α-synuclein remains unknown. A similar narrative as in AD has unfolded over the years: a native synaptic protein of mysterious function, various misfolded/aggregated/secreted species ranging from oligomers to fibrils, but no consensus on which is most important, if indeed there even is a single toxic species. The next frontier will be to answer this question, Frasier said. To address it, MJFF is funding mass spectrometry research to compare the forms of the protein detected by different commercial assays. This may help pin down post-translational modifications associated with toxicity, Frasier said.—Madolyn Bowman Rogers and Gabrielle Strobel
At AD/PD Meeting, New BACE Inhibitor Struts Its Stuff
Striving to be noticed in the hoopla over Biogen’s Phase 1 aducanumab data, other investigational therapies presented clinical data at the 12th International Conference on Alzheimer’s and Parkinson’s diseases, held March 18 to 22 in Nice on France’s Cote d’Azur. Consider JNJ-54861911. As recently as two years ago, a once-a-day drug that cuts CSF Aβ levels near to the ground in Phase 1 would have been big news. But by 2015, the field is not only replete with β-secretase inhibitors, but also enraptured—at least at the moment—by an antibody that teased an audience far beyond this research conference with hints of potential efficacy (see Part 1 of this series).
Yet the new BACE inhibitor is well worth noting. For one thing, some of its competition has since fallen by the wayside (e.g., Feb 2015 news, RG7129). For another, in offering details on the compound’s performance thus far, its developers showcased how the science of evaluating this type of drug is evolving. In Nice, Johannes Streffer of Janssen Research and Development in Beerse, Belgium, described the behavior, in the plasma and CSF of healthy volunteers, of a BACE inhibitor Janssen had licensed in 2012 from the Japanese pharmaceutical company Shionogi. Streffer’s talk afforded a peek at how early human trials nowadays are using continuous CSF monitoring and beginning to look more broadly at APP cleavage products beyond Aβ40 and 42.
Hit me! Yet another attempt at whacking BACE, a key drug target in Alzheimer’s therapy development, has entered clinical trials.
Streffer and colleagues evaluated this molecule in volunteers of the same age range as Alzheimer’s patients who were willing not only to take an unproven drug, but also to live for 36 hours with a catheter implanted in their back so the researchers could sample their CSF frequently and learn how the levels of certain biomarkers change over time. For a single-ascending-dose study, people wore the catheter for the whole study. For a second, multiple-ascending-dose study that lasted two weeks, volunteers agreed to a lumbar puncture at baseline followed by a 36-hour CSF catheter at the end of the study. The goal was to learn about the drug’s safety and tolerability, its reach into plasma and CSF, and its target engagement as measured by various effects on APP processing.
On safety—a closely watched subject for BACE inhibitors—Streffer said what most BACE inhibitor developers have said early in Phase 1, that is, no worrisome side effects related to the drug are yet in sight. Two people vomited, but most side effects were a consequence of the lumbar catheter and all were mild or moderate. In the studies Streffer reported at AD/PD, 94 people were exposed to the drug. “Overall, the inhibitor was very well tolerated,” Streffer told the audience. Unlike therapy with anti-Aβ antibodies, the participants’ ApoE genotype had no influence on safety or any outcome measured thus far for this small-molecule drug, Streffer reported.
JNJ-5486911 shoots into the plasma fast, reaching its peak concentration after one to two hours and washing out over nine to 16 hours. In the CSF the curve is slower, peaking at two to three hours and waning over the course of 36 hours. Importantly, drug levels in CSF are proportional to those in plasma, and dose-dependent, Streffer said. In people who took their randomized dose for two weeks, the drug reached a steady-state level after about a week. Studying a drug’s concentration in the body over time is important so researchers know it neither accumulates to dangerous levels nor gets metabolized to toxic byproducts before it reaches its target.
To measure what the drug does to APP, the scientists used an electrochemiluminescence-detection platform by Meso Scale Discovery, one of the companies competing with Roche Diagnostics and Fujirebio Europe (formerly Innogenetics) to make more robust assays for CSF analysis. On it, they used their own antibodies to quantify Aβ1-37, Aβ1-38, Aβ1-40 and Aβ1-42 simultaneously. They also used a combination of in-house and commercial MSD antibodies to quantify sAPPα, sAPPβ, and sAPP total in the presence of inhibitor. This set of tests captures aspects of what happens to BACE’s substrate when the enzyme is blocked. It includes some known Aβ species, though not the whole range that has emerged in recent mass spectrometry work, such as Aβ1-16, Aβ1-43, or N-terminally extended or N-terminally truncated or pyroglutamated species. Streffer said that reagents for some of these other species were in hand and would be studied, as well.
Looking first at the most abundant type of Aβ, the 1-40 peptide, Streffer reported that plasma and CSF are a bit different. In plasma, Aβ40 goes down in lockstep with inhibitor concentration going up. The reduction is dose-dependent and lasts longer than 36 hours after a single dose, Streffer said. Lower doses—in the range the company will take forward—reduced Aβ40 for about a day. The CSF showed a delayed and more protracted reaction, with Aβ40 concentrations going down dose-dependently after about eight hours. Streffer related the time course of the inhibitor’s pharmacodynamic response to the time course of newly produced Aβ in the brains of healthy people as measured by the SILK method (Mawuenyega et al., 2013), and concluded that the drug inhibits production of Aβ in the brain.
At the end of two weeks on the inhibitor, plasma and CSF Aβ40 stayed where they had been for another 24 to 36 hours: 5 mg cut CSF Aβ40 in half, and the 30, 50, and 90 mg doses pushed it below 20 percent. Claiming that his team has focused intense effort on eliminating variability from its CSF measurements, Streffer showed spaghetti graphs—flat lines of Aβ40 levels below 20 percent of baseline—from five study participants who had taken 50 mg of inhibitor for two weeks. “We have very stable CSF Aβ measurements now,” Streffer said. Variability at many levels—between centers, manufacturing lots, and test-retest of the same sample—has bedeviled CSF analysis in AD research for years. This appears to be on the verge of changing with the advent of new assays (see upcoming story in this series).
Like most drug developers these days, Janssen draws design help for larger trials from a quantitative pharmacokinetics model, essentially an in-silico oracle that puts Phase 1 data through a set of mathematical algorithms and draws a dose-response curve, among other outputs. In response to audience questions, Streffer replied that no one truly knows yet how much Aβ reduction is necessary for a clinical benefit, but his company picked 10 and 50 mg for their latest Phase 1b and 2a safety studies in early Alzheimer’s. “Even 25 mg achieves a solid 80 percent reduction of Aβ,” Streffer said.
How this BACE inhibitor is being developed stands in contrast to the strategy behind Biogen’s antibody BIIB037/aducanumab. The latter underwent two Phase 1 studies in a total of about 230 people, and skips Phase 2 to enter Phase 3 later this year. For JNJ-54861911, clinicaltrials.gov lists 10 Phase 1 studies. Its first Phase 2 trial tests safety plus similar dose, exposure, and target engagement biomarkers, as summarized in this story, for a six-month course of the drug; it makes no mention of cognitive or clinical efficacy at all. For a summary of trials on this compound, see JNJ-5486911.
After BACE has cleaved APP, its product C99 undergoes sequential processing by γ-secretase that yields not only Aβ40, but also longer and shorter versions of the peptide. Basic researchers have long argued that drug developers should know how all of them change, not just one or two. For example, Bart De Strooper of KU Leuven, Belgium, has called for an unbiased, comprehensive accounting of all cleavage products with mass spectrometry before taking a potential Alzheimer’s drug into expensive, high-profile trials. That would catch any potentially toxic APP byproducts early on, before they could trip up an investigational therapy. “If you inhibit one enzyme, another will take over,” De Strooper told Alzforum. Pharma companies tend to resist collecting more data than they think they need or can explain, or to adopt scientific methods they consider research-grade.
That said, Streffer’s team did branch out beyond studying Aβ40 and 42. At AD/PD, he reported that Aβ37, 38, 40, and 42 were reduced equally in CSF. What happens to APP when BACE is blocked? To address this question, the scientists profiled sAPPβ, the other product of APP cleavage by BACE besides C99; sAPPα, the product of APP cleavage by α-secretase, and sAPP total. At any given inhibitor dose, Aβ40 and sAPPβ dropped down to identical levels, Streffer said. This would be expected if both arise one-to-one from APP cleavage blocked by this compound. The concentration of sAPPα rose with increasing inhibitor doses, up to about 2.5-fold. sAPP total stayed unchanged at all doses. To Streffer’s mind, the decrease of sAPPβ was “very reassuring,” and the increase of sAPPα was “not concerning for the therapy.” sAPPα is thought to be non-toxic.
APP processing is drawing renewed interest in the field. Researchers continue to hunt for additional enzymes they suspect might cleave APP when BACE is out of commission. This, some believe, could generate additional fragments that warrant further study.—Gabrielle Strobel
D-peptides as Drugs? Protein Therapy Approaching Phase 1 Trials
An unusual, oligomer-morphing peptide therapy may soon graduate from preclinical studies into human trials for Alzheimer’s disease. Comprising D-amino acids, the peptides resist digestion by proteases that would make quick work of their chiral doppelgangers, the naturally occurring L-enantiomers. Over the past seven years, researchers led by Dieter Willbold at Forschungszentrum Jülich in Germany have been reporting that a D peptide called D3 converts Aβ oligomers into larger non-amyloidogenic species, reduces plaques in mouse brain, and improves cognitive function in several AD models. At the 12th International Conference on Alzheimer’s and Parkinson’s diseases, held March 18 to 22 in Nice, France, researchers from Willbold’s group presented new data on better-performing D3 derivatives that, like the original peptide, enter the brain efficiently when fed to mice. Barring unforeseen problems in ongoing toxicology and pharmacokinetic studies, Willbold said a D peptide is slated to enter human trials in 2016.
Sample peptide in the standard L form and its D mirror image.
Because D-enantiomers resist digestion in the stomach and are only weakly immunogenic, they are candidates for oral protein-based therapies. In 2008, Willbold and colleagues used a novel technique called mirror image phage display to find D3—a D-amino acid dodecamer that bound to Aβ fibrils. The scientists have since characterized it further (see van Groen et al., 2008; Aug 2009 conference story; Funke et al., 2010; and van Groen et al., 2012).
With their sights set on clinical development, the Willbold group synthesized D3 derivatives and tested them in vitro and in mice. These are the results they presented in Nice. In a talk and poster, respectively, his colleagues Oleksandr Brener and Tina Dunkelmann reported that one new compound, called C-1, targeted Aβ oligomers more efficiently than D3. The researchers added C-1 to a mixture of monomers, oligomers, fibrils, and larger species of Aβ, used density gradient centrifugation, and found that the oligomers disappeared while amounts of the larger fractions increased. Willbold said that the larger species formed in place of the oligomers did not go on to aggregate.
With collaborators Inga Kadish and Thomas van Groen from the University of Alabama at Birmingham, the researchers then compared the compounds head-to-head in two mouse models. After four weeks of intraperitoneal infusion with a minipump, both D3 and C-1 similarly boosted performance on the novel-object recognition test in the APPSweDI mouse model compared to untreated mice. TBA2 mice, which express a pyroglutamate Aβ peptide and develop motor and cognitive deficits, maintained their ability to balance on a spinning rod after one month’s treatment with either peptide, while C-1 preserved neural reflexes better than D3 did. Willbold told Alzforum that, to his mind, these experiments suggest that the peptides spare motor neurons from degeneration in this model. “The new derivatives work in some regards better, and in all regards at least as well as D3,” Willbold said. Other derivatives also outperformed the original D3, as presented on posters by Tamar Ziehm and Antonia Klein of Forschungszentrum Jülich.
The researchers next investigated the pharmacokinetic profile of D3 and its derivatives. They labeled D3 with radioactive tritium and delivered it into mice via several routes orally, intravenously, or by injecting it into the abdominal cavity. As presented on a poster by Nan Jiang, D3 had at least a 24-hour half-life in plasma, which is excellent compared to the hour-long half-life of typical L-peptides, Willbold said. The peptide easily passed the blood-brain barrier. Leonie Leithold’s poster reported that the derivatives of D3 had similar pharmacokinetics. Both Jiang and Leithold also work with Willbold.
“The derivatives of D3 are promising because of their direct interaction with Aβ oligomers. The bottom line is that at least in the transgenic mouse models, they have reproducible and promising effects,” commented Manfred Windisch of NeuroScios in Graz, Austria. He was not involved in the studies but has worked with D3 in the past.
The scientists are now completing preclinical toxicology and safety studies. Willbold said he has secured funding from the Helmholtz Validation Fund to initiate a Phase 1 trial. He would not reveal which D3 derivative has been picked, but expects the trial to start in 2016.—Jessica Shugart
New Genetics Frontiers: Finding Modifiers, Making Sense of Pathways
The laundry list of potential Alzheimer’s risk genes that have turned up in genome-wide association studies explains but a fraction of the total genetic burden of the disease, so geneticists are turning to whole-genome and exome sequencing to hunt down more suspects. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, speakers reported on the latest progress in this area, including a potential modifier gene identified in a large Colombian kindred with early onset disease. In parallel, geneticists are moving beyond listing genes. They are now analyzing genetic interactions to determine the underlying metabolic pathways most affected by each disorder. This provides clues as to why particular neuronal populations succumb in different neurodegenerative conditions, said John Hardy of University College London. Based on data from a number of such diseases, Hardy proposed specific metabolic processes that break down in different neuronal subtypes. “I hope that understanding selective vulnerability will be the first step to being able to compensate for it,” he told the audience.
For Alzheimer’s disease, the 30 or so loci identified to date account for about 30 percent of the genetic risk, Julie Williams of Cardiff University, U.K., said in her talk. GWAS have found mostly common genes that add little risk, so much of the missing heritability may lie in rare genes with larger effects. To find them, researchers are mining the genomes of families with inherited disease. Even when the causal mutation is already known, this approach can reveal separate factors that bring on disease earlier or later, said Kenneth Kosik of the University of California, Santa Barbara. In the Colombian kindred who carry the E280A PS1 mutation, most carriers develop mild cognitive impairment as defined by MCI criteria by the age of 44, and full-blown dementia five years later (Acosta-Baena et al., 2011). However, a few carriers have long intrigued researchers because they break the pattern, succumbing either much earlier or much later.
Kosik wanted to find out which genes are behind this. He analyzed whole-genome sequences from 117 members of this kindred. That huge dataset will likely reveal more insights, but even a first analysis turned up a protective haplotype of 56 kb that delayed disease onset by about 10 years. The haplotype was common in the kindred, occurring in about one in four people. It spanned a region of cytokine genes, and contained 22 SNPs that associated with delayed onset. Only one of these fell within the coding region of a gene, however. This SNP, rs1129844, marked eotaxin-1, also called CCL11.
Eotaxin. Good guy? Bad guy? Genetics raises the question.
Eotaxin has already made a name for itself in aging research. The concentration of this cytokine rises with age in both mice and people. It is one of the potentially deleterious aging factors identified in parabiosis research, in which young mice receive old blood (see Nov 2009 conference news; Aug 2011 news). In those studies, eotaxin suppressed the birth of new neurons and impaired learning.
The SNP Kosik found changes an alanine to a threonine at position 23, precisely where a signal peptide is cleaved from eotaxin, he noted. This site may also play a role in allowing the cytokine to bind its receptor, CCR3. Kosik wondered if the SNP might represent the functional mutation that confers protection. Cells transfected with the variant pumped out more eotaxin, suggesting a functional effect, though its direction surprised the researchers. “We might have expected a decrease in eotaxin secretion from a protective allele,” Kosik said.
Would this SNP protect other populations as well? To try to confirm the findings, Kosik collaborated with Aimee Kao at the University of California, San Francisco, to analyze 152 AD patients there. In this group also, the protective haplotype associated with later disease onset. Moreover, in people with this haplotype, eotaxin levels did not rise with age. Kosik looked for other modifiers in this group, and found an SNP in the IL4 receptor that was enriched in people who developed disease late, and absent in those who succumbed early. This SNP delayed disease by about seven years. It occurred only in people of Latino heritage. Intriguingly, ligands for IL4R can release eotaxin and induce Aβ clearance, Kosik said.
“We put forth the hypothesis that eotaxin release may mediate Aβ clearance epistatically with IL4R,” Kosik told the audience. Epistasis refers to an interaction between different genes, such that one modifies the expression or effect of the other. Low to moderate levels of eotaxin may be beneficial and higher levels deleterious, Koski suggested, adding “Having these SNPs may shift the curve toward the beneficial side.” However, he cautioned that these data are preliminary, as the populations tested were small and not powered to reach genome-wide significance. The finding needs to be confirmed in larger populations. So far, these SNPs have not shown up in GWAS results.
Other researchers are also analyzing genetic kindreds. Christine Van Broeckhoven of the VIB, University of Antwerp, Belgium, noted that even for familial AD, the causal mutation remains unknown in 90 to 95 percent of cases. She is leading genetic studies to try to pin down these genes. In a Belgium cohort of 343 familial AD cases, she found 52 separate mutations. Of these, 41 were in presenilin 1 or 2. Carriers had widely varying ages of onset, suggesting the presence of genetic modifiers, and she is looking for those now. Surprisingly, among the other 11 genes, loci linked to neurodegenerative diseases turned up frequently, in particular granulin, a risk factor for frontotemporal dementia (FTD). Some families carried more than one gene that segregated with disease; for example, one had both a PS1 mutation and a C9ORF72 expansion; the latter is linked to FTD and amyotrophic lateral sclerosis. How these additional mutations influence pathology in AD patients is unclear, Van Broeckhoven said. Neurogeneticists don’t yet know exactly what distinguishes a modifier that increases risk from a bona fide disease gene, or how common the presence of multiple disease-associated variants in a given person will turn out to be.
A complementary growing trend in genetics is that the same gene crops up in multiple diseases, for example TREM2 in both FTD and AD, and glucocerebrosidase (GBA) in both Gaucher’s and Parkinson’s, said Rita Guerreiro of UCL. This is called pleiotrophy, and exome sequencing is uncovering more such cases. By comparing affected and unaffected individuals within a family, exome sequencing can quickly home in on mutations. In 22 parent-child trios, Guerreiro’s group has found 26 confirmed de novo mutations. In one case, exome sequencing revealed that heterozygous mutations in polynucleotide kinase 3’-phosphatase caused familial ataxia in a Portuguese kindred (see Bras et al., 2015). Homozygous mutations in this gene, on the other hand, cause developmental delays, early seizures, and small brains. PNKP plays a role in repairing DNA.
As geneticists sort through associations, metabolic pathways have begun to emerge for each disease. Hardy pointed out that nearly every Alzheimer’s gene reported to date falls into one of four pathways: cholesterol metabolism, innate immunity, endosomal vesicle recycling, and protein ubiquitination. “I’d be suspicious of any new gene that didn’t fall into one of these pathways,” he said. In Parkinson’s disease, most genes are involved in lysosomal function, mitophagy, or immunity. “These pathways must mark those cellular systems that are susceptible to disease,” he suggested.
To explore this idea, Hardy looked at gene expression data for GWAS hits to find genes that are co-expressed. This analysis allowed him to define “modules” of co-regulated genes. In the case of ataxias, many GWAS hits mapped to one of two co-expression modules: calcium homeostasis or the ubiquitin proteasome system. Both of these co-expression modules occur only in the cerebellum, with the former active in Purkinje cells and the latter in granule cells (see Bettencourt et al., 2014). The motor symptoms of ataxia arise from perturbed cerebellar function. The data imply that the cells’ vulnerability arises from these particular systems. “We see two ways to get ataxia: to have disrupted calcium homeostasis or a disrupted ubiquitin proteasome,” Hardy said. Not only does each disease variant attack a different neuron type, it also has distinct clinical features, he added.
Using the same methodology, Hardy found similar patterns for other diseases, although the data were noisier. For Parkinson’s, many genes involved in mitochondrial complex I were co-expressed in dopamine neurons of the substantia nigra. They include Parkin, Pink1, and SNCA, as well as RAB39B, which was independently identified as a PD gene by another group (see Wilson et al., 2014). The finding suggests that mitochondrial function is crucial for dopaminergic neurons, and that stresses to this system precipitate PD. For dementias, including AD, FTD, dementia with Lewy bodies, and PD dementia, lysosomal gene co-expression modules turn up over and over, primarily affecting cortical pyramidal neurons, Hardy said. Meanwhile, many FTD and ALS genes, including C9ORF72, map to the ubiquitin proteasome module. These disorders target motor and cortical neurons, which may particularly rely on this waste disposal system.
“The overall suggestion of the work we’ve been doing is that each neuronal type, for reasons having to do with its function, is close to a catastrophic cliff,” Hardy said. Dysfunction in one of the genes involved in the particular task that puts the particular neuron type near that point can push the neuron over toward disease. “Selective vulnerability has to do with what catastrophic cliff the neuron is close to,” he concluded. Many genes in these modules are involved in cleaning up cellular damage, which may explain why the diseases only manifest with age, when accumulated wear and tear puts more pressure on neurons. In answer to an audience query, Hardy reiterated the idea that in many cases there may be a “second hit” that actually precipitates disease.—Madolyn Bowman Rogers
Wilson GR, Sim JC, McLean C, Giannandrea M, Galea CA, Riseley JR, Stephenson SE, Fitzpatrick E, Haas SA, Pope K, Hogan KJ, Gregg RG, Bromhead CJ, Wargowski DS, Lawrence CH, James PA, Churchyard A, Gao Y, Phelan DG, Gillies G, Salce N, Stanford L, Marsh AP, Mignogna ML, Hayflick SJ, Leventer RJ, Delatycki MB, Mellick GD, Kalscheuer VM, D'Adamo P, Bahlo M, Amor DJ, Lockhart PJ.
Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology.
Am J Hum Genet. 2014 Dec 4;95(6):729-35. Epub 2014 Nov 26
PubMed.
Changes in shape and structure can turn a well-behaved cellular protein into something altogether more sinister, but in many cases researchers do not know exactly which modifications are at fault. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, speakers grappled with what makes α-synuclein go rogue. Researchers led by Dennis Selkoe of Brigham and Women’s Hospital, Boston, correlated the presence of pathological variants of α-synuclein with a shift from multimeric forms toward freer monomers. The data address a controversy over whether α-synuclein can form higher-order structures that protect against aggregation. Meanwhile, Paola Picotti at ETH Zurich debuted a method of determining the aggregation state of α-synuclein in complex mixtures.
To study pathological changes, researchers first have to know the native state of α-synuclein. Scientists had long believed the molecule existed as a disordered monomer, but this view was challenged in 2011 when Selkoe and a group led by Gregory Petsko, who is now at Weill Cornell Medical College in New York City, reported the isolation of α-synuclein tetramers with an α-helical structure from red blood cells, neural cell cultures, and bacterial cells (see Aug 2011 news and commentary; Wang et al., 2011). Tetramers do not aggregate into amyloid-like fibrils, and might in fact represent the principal physiologic species, the researchers proposed. The results stirred debate, with other scientists reporting that they were unable to replicate the findings, albeit not using the same methods (see Feb 2012 news and commentary).
What Does Physiological α-Synuclein Look Like? Possible properties based on recent publications. [Image courtesy of Ulf Dettmer, BWH Boston.]
Since then, however, several other groups have independently turned up evidence for multimeric forms of α-synuclein (Trexler and Rhoades, 2012; Westphal and Chandra, 2013; Gould et al., 2014). Two independent groups, one led by Subhojit Roy at the University of California, San Diego, and the other by Nobel laureate Thomas Südhof at Stanford University, Palo Alto, California, reported that α-synuclein multimers corral and dock synaptic vesicles, suggesting a physiological role for the structures (see Oct 2014 news). In one of these synaptic studies (from the Südhof lab), multimers were described only on membranes, while cytosolic α-synuclein remained monomeric. New work from Südhof and first author Jacqueline Burré, who is also now at Weill Cornell, extends the findings, reporting that mutations in α-synuclein which interfere with membrane binding promote aggregation of the protein (see Burré et al., 2015).
In Nice, Selkoe emphasized that tetramers remain stable only in crowded molecular environments, falling apart upon cell lysis (see Dettmer et al., 2013; Luth et al., 2015). This may explain why most isolation methods fail to detect the higher-order structures, he said. At AD/PD, first author Ulf Dettmer said, “Alpha-synuclein multimerization is best studied in intact cells.” Dettmer described how he studied a human cerebral cortex biopsy removed from an otherwise healthy epileptic surgery patient. By finely mincing the material without lysing cells, and then adding a cross-linker to the intact brain bits, Dettmer was able to recover abundant α-synuclein tetramers. In his hands, these structures associated with cytosolic fractions, whereas monomeric α-synuclein appeared in both cytosol and membrane fractions. “We propose that an intact neuron maintains a physiological equilibrium of monomers and multimers,” Dettmer said.
In addition, the researchers traced the protein’s ability to form multimers to KTKEGV, six amino acids that repeat at least six times in the α-synuclein sequence. The known familial Parkinson’s mutation E46K replaces the glutamic acid in this sequence with lysine. Introducing this mutation lowered the amount of α-synuclein multimers present in cells, Dettmer said. Making the same mutation in a second of the repeat domains dropped multimers further, and three such mutations virtually abolished them, he added.
Does this relate to pathology? In a poster at AD/PD, Victoria von Saucken of Selkoe’s group showed that neuroblastoma cells expressing E46K-like and other mutations in the KTKEGV-repeat motifs accumulated aggregates of α-synuclein and died more quickly than control cells. Dettmer examined E46K and four additional missense mutations known to cause familial Parkinson’s disease: A53T, A30P, H50Q, and G51D. Like E46K, each mutation shifted the balance of α-synuclein in mouse brain from multimeric toward monomeric forms. Human neuronal cells expressing α-synuclein protein containing either a single or all five familial PD mutations built up more aggregated species due to tetramer destabilization, according to a poster presented by Tim Bartels of Brigham and Women’s.
The finding implies that stabilizing α-synuclein tetramers could help slow disease, Dettmer said in his talk. Similar strategies have been used to treat peripheral transthyretin amyloidosis (see Aug 2011 news; Dec 2013 news). One such drug, tafamidis, is currently approved for use in Europe and Japan, though not in the United States. Outside of this tetramer-stabilization approach, however, the debate about a native tetramer is not crucial to targeting pathologic, aggregated forms of α-synuclein, pharma researchers told Alzforum.
Acceptance of the multimeric model seems to be growing in the field. At AD/PD, a poster from Woon Ki Lim and Hae Ja Shin of Pusan National University in South Korea described a mass spectrometry analysis of α-synuclein that concluded that the protein forms highly ordered structures consistent with tetramers. The controversy now may be shifting to the questions of where multimers occur and what role they play. Another poster, from Harutsugu Tatebe and colleagues at Kyoto Prefectural University of Medicine in Japan reported no evidence of tetramers in human cerebrospinal fluid. To Dettmer’s mind, this finding is consistent with the tetramers being unstable outside of cells.
“The majority of data from many labs would suggest that in brain, soluble α-synuclein is monomeric but self-assembles on membranes,” Sreeganga Chandra at Yale University, New Haven, Connecticut, wrote to Alzforum. “However, it is possible to purify the tetramer from blood, though it is labile and will disassemble to monomers. So maybe this ‘controversy’ is just comparing brain versus blood.” The neuronal cell data from Dettmer et al. suggest otherwise. Chandra noted that she was not at AD/PD and could not comment on the latest data presented there.
“The data on α-synuclein multimers cannot be ignored anymore,” Roy wrote to Alzforum. “In particular, a concept is emerging that monomers and multimers are in dynamic equilibrium, and the multimeric state helps the protein associate with vesicles. It also seems that shifting this equilibrium away from multimers favors aggregation of the protein.”
Not everyone is convinced, however. “I don’t see evidence that α-synuclein in its native state exists predominantly as a tetramer, but we cannot rule out the possibility that it can form oligomers upon interaction with membrane,” Hilal Lashuel at the Swiss Federal Institute of Technology (EPFL), Lausanne, told Alzforum. He had not heard the latest talks. Others, including Südhof, see evidence for multimers, but not specifically for tetrameric forms. These scientists, too, have not yet seen the new data.
If the structure of α-synuclein determines the protein’s toxicity, researchers would want a quick and simple way to measure its state in body fluids. Picotti from the ETH said in her talk that an ideal method would allow researchers to analyze complex mixtures without labeling or purification. She developed a technique in which she first briefly digested mixtures with proteinase K before quenching the enzyme. This cuts only one or two sites on a protein, typically the most exposed, and the sites exposed vary depending on how the protein folds. Then the mixtures undergo tryptic digestion and mass spectrometry. Compared to standard mass spec results, the resulting spectra lack one or two peaks that correspond to the cleavage sites snipped by proteinase K (see Feng et al., 2014).
Picotti used this method to compare α-synuclein monomers to aggregated β-sheet forms. In each spectrum, she saw two unique sites that were missing in the other one. The unique sites served as markers for each conformation. “When we saw this, we got pretty excited,” she told the audience. The method may allow researchers to follow the kinetics of aggregation in vivo and to analyze modulating factors, she suggested. She told Alzforum she would like to apply the technique to the α-synuclein tetramers isolated by Selkoe’s group, as well, to see if her method can distinguish this species. In theory, the regions where subunits interface would be protected from proteolysis, potentially providing a unique signal from this form. She is also investigating whether the α-synuclein spectra could serve as a biomarker of disease in people. Other scientists said that this technique is innovative but not readily applicable for clinical purposes.—Madolyn Bowman Rogers
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ.
A soluble α-synuclein construct forms a dynamic tetramer.
Proc Natl Acad Sci U S A. 2011 Oct 25;108(43):17797-802. Epub 2011 Oct 17
PubMed.
The once-outlandish idea that misfolded proteins can propagate through the brain along anatomical connections has gone mainstream. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, no fewer than 30 talks and 50 posters addressed the topic of protein spread, with most scientists saying there is no doubt this occurs. Beyond that bit of consensus, however, little else is clear. Researchers debated which forms of different proteins constitute the pathological players, where in the body the process starts, and exactly how these species travel through the brain and pass between cells. Whether propagating proteins should be dubbed bona fide prions or are merely “prion-like” has become a semantic point of contention.
“It’s a sticking point for many in the field, because prions were originally defined as infectious particles. I think this nomenclature issue has impeded acceptance of the concept,” Lary Walker at Emory University, Atlanta, told Alzforum.
In the last few years, numerous animal studies have shown the spread of misfolded, aggregated forms of tau, Aβ, and α-synuclein to connected brain regions (e.g., Jun 2009 news; Jul 2009 news; Nov 2012 news). In many of these experiments, the seeds came from patients’ brains, emphasizing the capacity of neurodegenerative aggregates to corrupt native protein as prions do. Postmortem and PET imaging studies suggest the same propagation process occurs in human brain (for review, see Brettschneider et al., 2015). Other studies have turned up evidence in human brain of distinct strains of tau and Aβ—another prion-like feature (see Aug 2013 conference news; Sep 2013 news).
Seeds of Destruction.
Exogenous α-synuclein (green) seeds aggregation of endogenous protein (red), forming large deposits (overlay appears gold). [Image courtesy of Ronald Melki.]
What Seeds Disease?
But what exactly are these seeds, and how do they propagate? In Nice, Paul Fraser of the University of Toronto recapped the two classic models for prion-like transmission: template-directed refolding, and seeded nucleation (see Aguzzi and Calella, 2009). In one, a single misfolded protein molecule acts as the infectious agent and directs the misfolding of normal proteins one by one. The other model posits that a certain number of misfolded molecules always exist in equilibrium with healthy protein, but are harmless by themselves. It is only when a number of misfolded proteins assemble into an ordered structure that disease begins. These aggregates then recruit additional misfolded monomers, forming large protein deposits. Because the initial seeds form slowly, there is a long lag before disease begins, whereas subsequent recruitment occurs rapidly, causing pathology to run rampant through the brain. Claudio Soto of the University of Texas, Houston, believes this latter model has become accepted.
Seeds, then, are larger than monomers, but researchers disagree on how much larger. Are they oligomers? Fibrils? What initiates pathology? In the past decade, oligomers have drawn attention as a likely toxic entity, but Charles Duyckaerts of Pitié-Salpêtrière Hospital, Paris, described his recent study in which Aβ oligomers injected into mouse brains diffused widely within 24 hours (in press at Neurobiology of Aging). If soluble oligomers seed plaques, the plaques should then appear throughout the brain rather than only in regions connected to the injection site, he pointed out. He added that injected oligomers are quickly cleared by microglia, leaving no lasting damage. Soto disagreed with Duyckaerts’ interpretation that oligomers therefore do not seed disease, noting that injected prion proteins also disappear for a time before disease crops up.
To many researchers, the likely villains are fibrils. In cell culture studies, 4 nM of yeast prion fibrils are more toxic than 1 μM of oligomers, said Ronald Melki of the French National Center for Scientific Research, Gif–sur–Yvette, France (see Bucciantini et al., 2012). Patrik Brundin, of the Van Andel Institute in Grand Rapids, Michigan, noted that after injection into the olfactory bulb, pure preparations of monomers and oligomers travel efficiently between neurons but get swept up by microglia, whereas longer fibrils tend to get stuck outside cells; thus, neither form seeds well. In mouse experiments from Virginia Lee and John Trojanowski at the University of Pennsylvania Perelman School of Medicine, Philadelphia, sonicating α-synuclein fibrils to produce a mix of long and short strands produced the most efficient seeding, Brundin said. After a lag period of one to three months, this material triggered α-synuclein deposits in brain regions two synapses away from the injection site, as well as behavioral deficits (see Luk et al., 2012). “Fibrils are the species that trigger long-term pathology, but how you treat the fibril affects the dynamics of incubation time and efficiency,” Brundin concluded.
Evidence for the existence of different fibril strains is growing. Fraser showed that fibrils can have distinct tertiary structures, for example straight or twisted, each with its own stability and incubation time. Melki characterized synthetic α-synuclein aggregated under two different conditions, finding that each aggregate behaved differently in terms of its toxicity and ability to seed (see Bousset et al., 2013). Each strain has a “molecular bar code,” Melki said, jokingly comparing their shapes to different pasta, such as spaghetti versus linguini.
Posttranslational modifications may help determine how proteins aggregate into strains. Dietmar Thal of Ulm University, Germany, noted that phosphorylation and acetylation of Aβ and tau boost the number of intermediates and fibrils. The next question is to find out what forms lurk in the brains of patients. “We need to recover material from human brain and amplify it in vitro,” Melki said.
Does Parkinson’s Begin in Gut?
Deposits of α-synuclein (red) accumulate in enteric neurons (green), but not other cell types (nuclei in blue), in the gut of young transgenic α-synuclein mice. [Image courtesy of Markus Britschgi.]
Can aggregates seed disease from outside the brain? Some researchers have proposed that Parkinson’s disease could be triggered by α-synuclein accumulation in the gut, which then travels to the brainstem and midbrain (see Jul 2011 news series), or in the nose. At AD/PD, Markus Britschgi of Roche presented new data supporting the hypothesis that certain intestinal inflammations could kick off the disease (for a review, see Lema Tomé et al., 2013). Britschgi induced colitis in transgenic mice that expressed mutant human α-synuclein by putting dextran sodium sulfate in their drinking water for five days. In that time, intracellular aggregates of α-synuclein in enteric neurons roughly doubled, Britschgi said. To study longer-term effects, he induced chronic colitis in the mice for four weeks, then examined enteric neurons two months later. Although the colitis had long since resolved, the α-synuclein deposits persisted.
Many of these deposits were in macrophages. More gut α-synuclein accumulated in mice that lacked fractalkine receptor CX3CR1. This signaling pathway is known to be important for proper microglia-neuron interaction in the brain, and may likewise facilitate communication between macrophages and neurons in the gut, Britschgi said (see Nov 2011 news; Cardona et al., 2006). When these immune cells become activated by pathogens, environmental factors, or persistent deposits, they may damage gut tissue and further stress neurons, he suggested. Next Britschgi will investigate whether α-synuclein deposits eventually turn up in the brains of treated mice. Similar processes may occur in people, Britschgi noted. In eight out of 11 people with inflammatory bowel disease, Britschgi found α-synuclein deposits in gut macrophages and neurites. On the flip side, at least one study shows that people with Parkinson’s often suffer from inflammation of the colon (see Devos et al., 2013). A major genetic risk factor for PD, LRRK2, also predisposes to inflammatory bowel disease. If intestinal inflammation does precede Parkinson’s in some patients, it might provide an opportunity for early intervention, perhaps with antibacterial or anti-inflammatory agents, or even anti-α-synuclein antibodies, Britschgi wrote to Alzforum.
Some audience members countered that α-synuclein accumulation occurs in the guts of healthy controls, too, and that epidemiological studies to date have not reported a link between intestinal disease and Parkinson’s. Britschgi noted that this idea has been little studied, saying it would be worth looking for a connection in large epidemiology datasets.
Mechanisms Mostly Murky
Perhaps the greatest unknown about protein propagation is how it happens. First, here is what’s clear: Researchers agree that protein aggregates travel along anatomical networks, spreading to connecting rather than just neighboring regions. Both aggregated Aβ and tau progress through the brain in an anterograde course, or down axons, said Thal. The initial site of seeding determines which neuroanatomical pathways and networks are affected, speakers concurred. For example, tau injections in the hippocampus travel to the piriform cortex, while cortical injections sprout deposits in the hippocampus, amygdala, and midbrain, reported Diederik Moechars of Janssen Pharmaceutica, Beerse, Belgium (see Peeraer et al., 2015). Seeding of different regions determines the behavioral outcomes as well, and might be responsible for heterogeneous symptoms seen in patients, noted Ilse Dewachter of the Université Catholique de Louvain, Brussels, Belgium.
α-Synuclein Spread.
Aggregates of α-synuclein (green) spread into the anterior olfactory nucleus (pictured, nuclei in blue) after injection of fibrils in olfactory bulb. [Image courtesy of Nolwen Rey.]
To illustrate that anatomical connections are vulnerable to spread, Duyckaerts described a case of a woman who died at 85 from Alzheimer’s. A small piece of her cortex had been disconnected from the rest for 20 years, when a brain surgery severed it. In contrast to the rest of her brain, this isolated region contained no neuritic plaques or neurofibrillary tangles, only diffuse Aβ deposits. This finding highlights that the spread of pathology requires connection, Duyckaerts said.
Beyond this broad picture, however, details are fuzzy. “There is very little mechanistic data about how the process works,” Marc Diamond at the University of Texas Southwestern Medical Center, Dallas, wrote to Alzforum. How are aggregates transported inside neurons? In cell culture studies, researchers have tracked aggregates moving both anterogradely and retrogradely along axons via fast transport, in other words, carried by molecular motors along microtubules, Melki said (see Freundt et al., 2012). However, Thal noted that this remains to be proven in human brain. Aggregates might instead move along the outside of axons by guided diffusion or glial transport, he suggested.
Even less clear is how aggregates might enter and exit cells. First, about getting in. In 2013, Diamond reported that neurons take up tau and α-synuclein aggregates through a form of endocytosis, after tau sticks to heparan sulfate proteoglycans on the cell surface (see Mar 2013 conference news). To date, no replication studies have been published, nor are there papers proposing a detailed entry mechanism. At AD/PD, researchers led by Pamela McLean of the Mayo Clinic in Jacksonville, Florida, reported on a poster that α-synuclein oligomers inside exosomes were taken up equally well by cells that lacked HSPGs and cells that had them, suggesting that these molecules are not always necessary for endocytosis. Free α-synuclein, on the other hand, remained stuck to the outside of cells. Another poster, from Eliezer Masliah and colleagues at the University of California, San Diego, reported the opposite. In their hands, internalization of both oligomeric and fibrillar α-synuclein required the presence of functioning HSPGs on the cell surface. The reason for the discrepancy is not yet clear. Complicating matters, many researchers suggest that different types of aggregate may use distinct methods to infiltrate cells.
How do seeds get out? Researchers believe that neurons spit out toxic aggregates packaged in small membraneous vesicles called exosomes. Existing data indicate that only a small fraction of protein secretion occurs in these structures, but exosomes may still be crucial to the process, Brundin said. He found that lipids in these vesicles accelerated aggregation of α-synuclein fibrils in vitro (see Grey et al., 2015). Many researchers are currently probing the role of exosomes in neurodegenerative disease (see Dec 2014 conference news). Two posters at AD/PD reported evidence for transfer of Aβ aggregates inside exosomes. In one, researchers led by Efrat Levy at the Nathan S. Kline Institute for Psychiatric Research, Orangeburg, New York, described worse amyloid pathology in young Tg2576 mice injected with exosomes from older Tg2576 animals, but not in young Tg2576 mice that received exosomes from aged control mice. In the other, Martin Hallbeck and colleagues at Linköping University, Sweden, tracked the fate of fluorescently labeled Aβ oligomers in a neuronal culture system. They found Aβ inside secreted exosomes, which was taken up by other neurons through endocytosis.
Exosomes are not the only vessels in which seeding aggregates might escape cells. There are also ectosomes—larger vesicles than exosomes that get pinched off directly from the plasma membrane. They are plausible vehicles for tau, a cytosolic protein not associated with the secretory pathway, said Morvane Colin at AD/PD. Colin collaborates with Luc Buee at University of Lille, France, on mechanisms of tau spread. Colin last year had reported physiological release of tau ectosomes from cultured cells and detected them in the brain interstitial fluid of rats (Dujardin et al., 2014). At AD/PD, Colin expanded on that work. She reported that she found increased levels of tau ectosomes in the brain interstitial fluid of a new transgenic rat model of tauopathy, as well as in a hippocampal injection macaque model of tauopathy. However, because most tau is in free form, it is unclear whether these vesicular forms are the pathogenic species, Colin told the audience. A poster from Jürgen Götz and colleagues at the University of Queensland, Australia, reported that tau aggregates in ectosomes are differently phosphorylated than those in exosomes, hinting that there could be a difference in toxicity between these species.
What’s in a Name: The Prion Debate
What to call propagating proteins has become a hot-button issue. Prion researchers point out that Tau, Aβ, and α-synuclein aggregates pass most prion identity tests. Rattling off a list of characteristics, Soto noted that even some features he initially doubted have proven to exist.
For example, exposing animals to neurodegenerative seeds from various peripheral routes can stimulate brain deposition. Presenting data on peripherally administered Aβ, Rodrigo Morales in Soto’s group reported that eye drops were most efficient, followed by intraperitoneal, then intramuscular injection. Only eating Aβ aggregates failed to produce brain pathology. Morales reported that diluting Aβ aggregates delayed the appearance of pathology in linear fashion, as is the case with prion protein. The minimum amount of inoculum that produced disease was about 343 femtograms, similar to the minimum infectious dose of aggressive prion strains, he said. Mathias Jucker at the German Center for Neurodegenerative Diseases in Tübingen, Germany, previously reported that Aβ amounts as low as one femtogram could seed plaques (see Sep 2014 news).
The one test Aβ aggregates fail is that of person-to-person transmission under natural conditions, Soto said. The prion disease Creutzfeldt-Jakob also does not transmit between people through normal daily contact. Few studies have tried to measure human transmission of neurodegenerative disease, though a 2013 paper from Trojanowski and Lee reported that people who received human growth hormone from cadaver preparations were no more likely to develop Alzheimer’s or Parkinson’s than the general population (see Feb 2013 news). Researchers note that this study is not definitive, saying additional research should determine whether neurodegenerative diseases can be transferred through medical procedures under any conditions.
In fact, many researchers avoid the term “prion” to avoid stirring up confusion and fear (see Jun 2012 news). In a new review, Walker argues that redefining the term prion as a “proteinaceous nucleating particle,” rather than a “proteinaceous infectious particle,” would solve the issue. This would allow the term to encompass both prion protein diseases and neurodegenerative protein aggregates, Walker said (see Walker and Jucker, 2015). “As concepts evolve, definitions should evolve,” he told Alzforum.—Madolyn Bowman Rogers
The Feud, Act II: Do Alzheimer’s Genes Affect Amyloid or Tau?
Most GWAS hits only nudge the risk of developing Alzheimer's; their real power lies in what they can tell researchers about how the disease develops. For some, scientists are starting to decipher how the proteins might influence pathology and—why, hello!—the old rivalry between Aβ and tau resurfaces. (In truth, the religious war of yesteryear lives on only as fodder for some media stories; most scientists agree both are important.) But consider this case in point: Previous work on BIN1 has linked the protein to tau pathology, not to amyloid. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, however,Taisuke Tomita of the University of Tokyo presented new data that tied both GWAS hits BIN1 and PICALMto Aβ production via endocytosis.
Tomita found that in mammalian cell cultures, each protein affected the endosomal recycling of one of the two secretases that produce Aβ, potentially boosting cleavage of the peptide. Meanwhile, poster presentations at AD/PD strengthened the case for BIN1 directly interacting with tau and exacerbating its toxicity, though the mechanism remains unclear. Some studies have correlated BIN1 expression with tau tangles in human brain, hinting that this interaction is relevant in the disease. However, more work is needed to elucidate how these proteins affect AD risk. If past tauist-baptist feuds are any guide, researchers may well find that the answer turns out to be neither one nor the other alone, but a bit of both.
BIN1 and PICALM were two of the first AD loci identified by GWAS, and they have popped up consistently in independent samples (see Apr 2011 news; Oct 2013 news). Risk variants in both genes correlate with a three- to six-month-earlier diagnosis of AD, suggesting they hasten pathology (see Nov 2014 news). Both proteins promote clathrin-mediated endocytosis, leading many researchers to speculate that they could alter the processing of endosomal amyloid precursor protein (APP) into Aβ, but hard data for this has been lacking (for review, see Bohm et al., 2015).
Interrupted Journey.
In control cells (left panel), most BACE (green) resides on the cell surface or travels to lysosomes for degradation, with only a little in early endosomes (purple). In cells lacking BIN1 (right panel), BACE builds up in endosomes (overlay appears white), where it can chop APP into Aβ. Bar, 10 μm. [Image courtesy of Taisuke Tomita.]
To pin down a mechanism, Tomita and colleague Takeshi Iwatsubo at the University of Tokyo turned to BIN1 knockout mice. BIN1 is an adaptor protein that helps pinch off endocytic vesicles. In the knockouts, Tomita saw higher levels of Aβ and BACE1. To find out what was happening to the secretase, he labeled cell-surface BACE1 in cultured neurons from the knockouts and tracked its movement. After 30 minutes, BACE1 appeared in early endosomes, just as it did in wild-type cells. After 90 minutes, however, BACE1 in the wild types had traveled to late endosomes and lysosomes for degradation, while in the knockouts the enzyme fell back, remaining stuck in endosomes. Since most Aβ is produced in endosomes after sequential cleavage of APP by BACE1 and γ-secretase, detaining BACE1 here would likely pump up Aβ production, Tomita noted. In addition, BACE1 might be recycled back to the plasma membrane, starting the process all over. The data add teeth to the idea that BIN1 can exert a direct effect on Aβ production by mediating BACE1 trafficking to the lysosome, Tomita concluded. He had previously presented some of this work at an Oct 2013 BACE conference (see Dec 2013 conference news).
Tomita is currently examining alternative splicing variants that confer a heightened risk of the disease. Preliminary results hint that one variant, which lacks the C-terminus, might act as a dominant negative, lowering the activity of normal BIN1. Other variants lie outside the coding region and might lower expression of the protein, he said.
This contrasts with other studies of BIN1, which point to tau. Previously, Jean-Charles Lambert of Lille University, France, associated an AD risk allele of BIN1 with higher BIN1 expression and more tau pathology in human brains (see Chapuis et al., 2013). The findings dovetailed with another human postmortem study, in which higher BIN1 expression in several brain regions correlated with more tau tangles, but not with amyloid plaques. In that study, splicing of BIN1 in AD brains was shifted away from the longest isoform toward shorter isoforms (see Holler et al., 2014). In accord with this, missplicing of BIN1 occurs in myotonic dystrophy, a disease also characterized by tau tangles in the brain (see Fugier et al., 2011; Caillet-Boudin et al., 2014).
How might BIN1 affect tau? Lambert had previously reported a direct interaction between the proteins, as seen in pull-down assays (see Aug 2012 conference news). In Nice, he and first author Yoann Sottejeau extended these findings. In new studies, they combined pull-downs of different fragments of BIN1 and tau with nuclear magnetic resonance spectroscopy to analyze binding. This revealed that the SH3 domain of BIN1 binds the proline-rich domain of tau, at residues 211-231. Multiple phosphorylation sites associated with AD are located within the proline-rich domain, and could potentially affect this interaction, Sottejeau noted in his poster.
To parse how this interaction affects pathology, researchers are using flies. Previously, Lambert had reported that knocking down the BIN1 homolog Amphiphysin in a Drosophila tauopathy model made the flies healthier and more agile, in line with the human data suggesting that too much BIN1 worsens tau tangles (see Aug 2011 conference news). In Nice, however, Thomas Jahn of the German Cancer Research Center, Heidelberg, presented opposite results. In a poster, first author Nina Dräger described finding higher tau levels and a worsening phenotype after knockdown of Amphiphysin in tau model flies. This implied the adaptor protein helped control tau pathology. Moreover, expressing human tau in wild-type flies and neuronal cells stimulated Amphiphysin expression. This might represent a compensatory mechanism to modulate tau accumulation, the authors suggested. It is unclear why different fly models produced such divergent results. In contrast to the mammalian data, neither Lambert nor Jahn saw any effect of BIN1 knockdown on flies modeling Aβ pathology. However, Jahn noted that this finding does not contradict the cell culture results, as both fly models overexpressed the Aβ peptide, not APP, and thus were not designed to assess effects on cleavage.
What to make of these conflicting findings? “I think the data linking BIN1 to tau pathology are solid,” noted Steven Estus at the University of Kentucky, Lexington. “However, BIN1 has a number of interaction domains and several isoforms, so it could be involved in many processes.” Estus suggested it would be worthwhile to see if BIN1 polymorphisms turned up in other tauopathies, such as frontotemporal dementia or progressive supranuclear palsy. Tomita told Alzforum he is examining BIN1 mouse models for any effects on tau. He found no change in levels of endogenous mouse tau in BIN1 conditional knockouts. Next, he will cross the conditional knockouts with transgenic mice that model tauopathies.
PICALM Promotes Amyloid Production.
Aβ (brown) accumulates in the piriform cortex of APP transgenic mice (left panel), but not in littermates that express half the normal level of PICALM (right panel). Bar, 100 μm. [Image courtesy of Taisuke Tomita.]
For PICALM, on the other hand, the data seem to point more clearly toward an amyloid link. An earlier study by researchers at Washington University in St. Louis reported that PICALM’s protein product, CALM, helps internalize APP, facilitating its cleavage into Aβ (see Xiao et al., 2012). Previous work by Tomita complemented that finding. He reported a lower ratio of Aβ42 to total Aβ in heterozygous PICALM knockout mice, which correlated with sluggish endocytosis of γ-secretase (see Kanatsu et al., 2014).
In Nice, Tomita presented new work in this area. To see if PICALM could affect amyloid pathology, he crossed PICALM heterozygous knockouts with transgenic mice that overexpressed APP. The offspring deposited about a quarter as much amyloid as the APP parent did, he told the audience. The researchers then introduced a mutation in the ANTH domain of CALM that weakened the protein’s ability to bind membrane lipids, which is crucial for promoting endocytosis. Overexpressing this mutant in cell culture lowered the Aβ42 ratio, just as lack of the protein did. This again tied the effect to altered endocytosis.
When CALM was mutated, more γ-secretase loitered on the cell surface. This also happened after knockdown of PICALM in wild-type cells. The data strengthen the idea that CALM helps internalize γ-secretase into endosomes, where it can produce Aβ, Tomita concluded. Next, he investigated how a rare variant of PICALM, I34M, affects this process. The SNP occurs in the lipid-binding site of the ANTH domain, and was reported as a possible protective variant. The researchers found that this variant poorly bound γ-secretase, stranding the enzyme on the cell surface. In these cells, the Aβ42 to total Aβ ratio dropped. Together, the PICALM and BIN1 data suggest that subtle effects on Aβ production can hasten or delay disease, Tomita told the audience in Nice.
“Thanks to its role in clathrin-mediated endocytosis, it makes sense to tie PICALM to APP processing, but the jury is still out on what exactly it’s doing,” Estus told Alzforum. He pointed out that PICALM plays a crucial role in keeping endocytosis running smoothly. Homozygous knockout mice die at weaning. For this reason, the protein probably would not be a tractable drug target, he noted.
Moreover, as with BIN1, PICALM has been linked to tau pathology. A 2014 paper from researchers at the University of Cambridge reported that CALM promoted tau clearance by stimulating autophagy. Both too much and too little CALM dialed down this waste-disposal system in fly and zebrafish models, leading to tau accumulation (see Moreau et al., 2014). Other studies are pursuing different mechanisms. PICALM is highly expressed in the vasculature, leading some researchers to speculate it might play a role in clearing Aβ from brain.—Madolyn Bowman Rogers
Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N.
Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy.
Nat Med. 2011 Jun;17(6):720-5. Epub 2011 May 29
PubMed.
Could Adaptive Immunity Set the Brakes on Amyloid?
In the last few years, neuroinflammation has surged to the forefront of Alzheimer’s disease research. In dozens of presentations at the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, researchers grappled with the complexities of how the immune system can both speed and slow disease progression. At this point, there are more questions than answers, scientists agreed. One emerging theme was an unexpected involvement of the adaptive immune system in AD, supported by genetic and animal studies. Researchers also debated, again, what role peripheral macrophages play in cleaning up the brain. Others focused on microglia, trying to parse what makes these cells a force for good rather than ill in the diseased brain, and what role the risk factor TREM2 plays in this (see Part 10 of this series). Overall, the data at AD/PD generated excitement but no consensus.
“The talks represented excellent science, but we have a long way to go yet to arrive at a unifying theory of inflammation in AD,” Cynthia Lemere of Brigham and Women’s Hospital, Boston, told Alzforum, expressing a common sentiment.
Genetic studies have driven the recent interest in inflammation. In Nice, Rudolph Tanzi of Massachusetts General Hospital, Charlestown, added new data highlighting the role the immune system plays in disease risk. He presented recent results from the ongoing Alzheimer’s Genome Project, which now has sequenced whole genomes from 1,480 people in 437 families with inherited AD. Unlike GWAS, which find risk markers but usually do not identify functional variants, whole-genome sequences allow researchers to more easily home in on the polymorphisms that cause disease. These projects also generate a huge amount of data. In the case of the Alzheimer’s Genome Project, that amounts to about half a petabyte so far, or 500,000,000,000,000 bytes—and with that come technical challenges to clean up and analyze the findings, Tanzi noted. In preliminary analyses, the researchers focused on known GWAS risk loci. So far, they have turned up 180 rare variants in these genes that segregate with disease in families and are predicted to alter protein function. Many of the affected genes are involved in phagocytosis and chemokine signaling. “Immune genes are the fastest-growing set in AD,” Tanzi told the audience.
The researchers also looked for structural variations, such as duplications or deletions, that segregated with disease. They found three hits with genome-wide significance. Unexpectedly, all were genes involved in adaptive immunity: the T cell receptor α, the IgG heavy chain, and the IgG κ gene.
Adaptive Immunity Quashes Amyloid.
5xFAD mice lacking B and T cells (right) accumulate four times the volume of amyloid plaques (red) in their dentate gyrus, compared to mice with intact immune systems (left). [Image courtesy of Samuel Marsh and Mathew Blurton-Jones.]
How might adaptive immune genes influence AD? Tanzi speculated that defects in this system could leave the brain more vulnerable to infiltration by pathogens such as the candida fungus and herpes simplex virus (see Feb 2011 webinar). One previous study proposed that a normal function of Aβ is to attack microbes, and that it gets released in response to infection (see Mar 2010 news). Tanzi proposed an immunogenetic hypothesis of Aβ, in which genetic variants first compromise immune function in the brain, which over time leads to low-grade infections. In response, neurons might pump up Aβ production, kicking off pathology and eventually leading to plaques and tangles. Tanzi is pursuing this hypothesis with Robert Moir at MGH. In preliminary studies, they have found that injecting salmonella bacteria into the hippocampus of 5xFAD mice rapidly seeds amyloid plaques, Tanzi said.
To date, few researchers have studied effects of the adaptive immune system in AD, since B and T cells are confined to the bloodstream and are believed to stay outside the blood-brain barrier under most circumstances. One researcher who has focused on this has been Michal Schwartz at the Weizmann Institute of Science in Rehovot, Israel. She has long championed a role for the peripheral immune system in neurodegenerative disease. Her previous studies hinted that T cells may influence brain microglia via cytokines and aid in injury repair (see Jul 2006 news; Sep 2009 news). In Nice, other labs took up the cause, emphasizing the importance of adaptive immune cells in AD. These talks received an enthusiastic response from the audience, who peppered speakers with questions.
Samuel Marsh works with Mathew Blurton-Jones at the University of California, Irvine. He made a case for adaptive immunity controlling AD pathology. Marsh became interested in this when he crossed 5xFAD mice with a transgenic strain missing B and T cells in order to facilitate stem cell studies. The latter mice, Rag2-/-/IL2Rγ-/-, lack two genes essential for the generation of lymphocytes (see Mazurier et al., 1999). Marsh noticed that the Rag-5xFAD offspring had twice as much soluble Aβ40 and Aβ42 and up to four times the volume of amyloid plaque in their brains as the 5xFAD controls. Looking for the mechanism, Marsh found increases in pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα in their brains. The hybrids also had slightly more activated, Iba+ microglia in their dentate gyrus. The results suggest that adaptive immune cells communicate with the CNS and affect inflammation there, Marsh concluded. Another possibility is that antibodies normally cross the blood-brain barrier and aid amyloid clearance, he suggested. He is now transplanting bone marrow from young and old mice into these animals to find out if this will ameliorate pathology.
How might the adaptive immune system communicate with the brain? Perhaps through effects on peripheral phagocytes, suggested Kuti Baruch, who works with Schwartz. Previous studies reported that peripheral monocytes can infiltrate brain tissue to mop up amyloid, taking over cleanup duties from sluggish microglia (see Jun 2008 news; Apr 2011 news; Aug 2011 news). In earlier work, Schwartz and colleagues found that the choroid plexus tissue that lines the brain’s ventricles expresses chemokines and adhesion molecules that attract monocytes to sites of injury and help them enter the brain (see Shechter et al., 2013). Expression of these trafficking molecules depends on interferon-γ released by memory effector T cells that cozy up to the choroid plexus epithelium (see Kunis et al., 2013). Aging alters this cross-talk between T cells and choroid plexus, shifting signaling from IFN-γ to IL-4 and creating a more pro-inflammatory milieu (see Aug 2014 news; Baruch et al., 2013).
In Nice, Baruch suggested that in AD, problems with the choroid plexus might hinder its recruitment of monocytes. To test this idea, he examined 5xFAD mice and indeed found less IFN-γ at the choroid plexus, as well as a steep drop in the expression of various leukocyte-recruiting molecules, such as ICAM1, VCAM1, CXCL10, CD73, and CCL2. To see if he could reverse this, Baruch injected 6-month-old mice weekly with glatiramer acetate (GA). This multiple sclerosis drug stimulates T cells. In treated animals, more monocytes infiltrated the brain, became macrophages and surrounded plaques. Inflammation dampened, plaques shrank, and behavioral test performance improved. This benefit lasted for up to a month after injection, Baruch said. He cautioned, however, that daily GA injections had the opposite effect, suppressing leukocyte recruitment and exacerbating inflammation. Future modulation of the immune system would have to be precisely calibrated to benefit patients, he concluded.
In a similar vein, a poster presented by Guillaume Dorothee of INSERM, Paris, emphasized the importance of T cells in modulating the phagocytes’ response in AD. Dorothee and colleagues depleted T regulatory cells in APPPS1 mice, and saw fewer microglia around amyloid plaques along with an earlier onset of cognitive problems. Conversely, when they selectively amplified the T regulatory cell population by treating with low-dose IL-2, more microglia surrounded plaques and cognitive deficits held off. Although the precise mechanisms of action of T regulatory cells are still under investigation, the data highlight the therapeutic potential of innovative Treg-based immunotherapy for AD, Dorothee told Alzforum.
Do T cells exert all their influence from outside the brain? Perhaps not. Maria-Teresa Ferretti, who works with Roger Nitsch at the University of Zurich, noted that several groups have reported T cells in the parenchyma of AD brains, although the idea remains controversial (for review, see Ransohoff and Engelhardt, 2012; Togo et al., 2002). Ferretti examined human postmortem brain samples, and found higher numbers of T cells in hippocampal sections from AD patients versus controls. To study this further, she turned to aged APP transgenic mice. These animals had about twice as many T cells in their brain tissue as did wild-type mice, she reported. How did they get in? T cells were not correlated with micro-hemorrhages, nor did they get stuck in blood vessels or perivascular space, Ferretti found. Instead, she saw an increase of adhesion molecules such as ICAM-1 and VCAM-1 in the brain’s blood vessels, and concluded that the endothelium in AD brains encourages entry of T cells. What the cells are doing is less clear. Ferretti saw little evidence that they proliferated. She analyzed their markers and found the cells expressed low levels of IFN-γ, suggesting they were not cytotoxic and instead might play a regulatory function.
Overall, AD/PD talks pointed toward a role for adaptive immunity in AD. “The adaptive process appears to be less robust than innate immunity, in terms of the number of immune cells in brain. But regardless, it has a major impact on the downstream events in pathogenesis,” noted Lemere in an email to Alzforum.
Other scientists added nuance to the idea that peripheral macrophages aid phagocytosis in the brain. In a poster, Stefan Prokop, Kelly Miller and Frank Heppner of Charité University Medicine, Berlin, described ablating microglia in 5-month-old APPPS1 mice through a genetic cross. In response, peripheral monocytes infiltrated and repopulated the brain. However, these new cells did not surround amyloid plaques, nor did they lower the Aβ burden. Even treating the mice with anti-Aβ antibodies failed to activate these macrophages. Additional triggers might be necessary to stimulate macrophages to chew up Aβ, the authors suggested.
Researchers disagree on the role of macrophages versus microglia in AD. One problem is that many studies distinguish the cell types using markers that are not always reliable, Todd Golde of the University of Florida, Gainesville, wrote to Alzforum. In ongoing studies, researchers are attempting to better delineate these immune cells (see Feb 2015 conference news). For the latest on microglial studies, see Part 10 of this series.—Madolyn Bowman Rogers
The brain’s resident immune cells, microglia, play a Jekyll-and-Hyde game in Alzheimer’s disease. Researchers at the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, outlined their latest efforts to discover what makes these cells good or bad. Some speakers talked about characterizing them in healthy and diseased brains. Others presented new data supporting TREM2’s role in promoting phagocytosis, although controversy continues over whether this receptor even marks microglia or infiltrating macrophages. Other talks pointed to the enzyme arginase-1 as a marker of helpful microglia, or implicated these cells in triggering Parkinson’s disease. As yet, no single clear picture has emerged; instead, accumulating data emphasize the complex and shifting nature of these cells.
Researchers used to divide microglia neatly between an M1 phenotype that promotes inflammation, and a more beneficial M2 phenotype that specializes in phagocytosis. This simple schema is being supplanted by the recognition that microglia can assume numerous states characterized by multiple overlapping markers (see Mar 2013 conference news).
In Nice, Delphine Boche of the University of Southampton, U.K., discussed efforts to more fully characterize healthy versus harmful microglia. She examined multiple microglial markers in postmortem brain slices from 148 dementia patients and 130 age-matched controls. Samples came from the MRC Cognitive Function and Ageing Study (CFAS). She then correlated the findings with cognitive scores on the Mini-Mental State Examination. Microglial expression of CD64 and Iba1 associated with high test performance, whereas CD68 and HLA-DR predominated in people who had scored low. CD64 is involved in activation and Iba1 in motility, while CD68 relates to phagocytosis and HLA-DR to antigen presentation, she noted. She saw high CD68, HLA-DR, and CD64 and low Iba1 in ApoE4 carriers as well, tying this risk factor to inflammation.
How did Alzheimer’s pathology affect the microglia’s phenotype? CD68 was linked to higher tau pathology, while CD64 showed up in brains with more diffuse plaques but less tau, Boche reported. This hints that CD64 might be in play at an earlier and CD68 at a later stage of AD pathology.
Boche saw a difference not only in microglia phenotype but also in overall activation. Brains with Alzheimer’s pathology had more activated phagocytes, but there was a sharp distinction between people with and without dementia. In those with normal cognition, AD pathology was associated with less or no microglial activation, but not more. “Microglia respond differently to Aβ and tau in people with and without dementia,” she noted. “Microglial status may be pivotal in AD pathogenesis.”
The Secret Life of TREM2. TREM2 is cleaved from the surface of microglia by ADAM10, freeing a soluble portion that may interact with ligands on neurons. Disease-associated mutations in TREM2 affect these processes. [Image courtesy of Daniel Sevlever.]
Boche stained for TREM2 in microglia as well, but reported that she saw the marker only on macrophages derived from peripheral cells. The finding fits with recent data from Bruce Lamb at the Cleveland Clinic, Ohio, who argued that TREM2-expressing cells in the brain come from the blood (see Jay et al., 2015). The issue has raised controversy among researchers in the field. “I think the data are convincing that TREM 2 is a protein expressed by infiltrating cells,” David Morgan at the University of South Florida, Tampa, wrote to Alzforum. Others disagree, maintaining that they do see TREM2 on resident microglia (see Dec 2014 conference news). One problem is that the different studies use varying methods. “Further studies using similar protocols, mouse models, and assays will be required to sort out the TREM2 story,” Cynthia Lemere of Brigham and Women’s Hospital, Boston, wrote to Alzforum.
TREM2 has drawn intense interest in AD research ever since geneticists reported that rare variants in this gene confer as much risk as the ApoE4 allele (see Nov 2012 news). Most researchers agree that these variants result in a loss of TREM2 function, implying that the protein helps control AD pathology, although Lamb recently reported fewer plaques in TREM2 knockouts (see Feb 2015 conference news).
In Nice, Christian Haass of Ludwig-Maximilians-Universität, Munich, argued that TREM2 aids phagocytosis. A single-pass transmembrane protein, similar in structure to amyloid precursor protein (APP), TREM2 is cleaved by the α-secretase ADAM10 and its outer portion shed into extracellular space. Haass previously reported that a TREM2 mutation associated with frontotemporal dementia, T66M, inhibited maturation and subsequent shedding of the protein because it got stuck inside endoplasmic reticulum. Both mice and people with T66M and other missense mutations had lowered levels of soluble TREM2 in plasma and cerebrospinal fluid (see Jul 2014 webinar; Kleinberger et al., 2014).
On the other hand, the R47H mutation associated with AD was being shed normally. In ongoing work, Haass is measuring soluble TREM2 in Alzheimer’s disease. At AD/PD, he reported high soluble TREM2 levels in the CSF of people with preclinical AD or mild cognitive impairment due to AD, compared with age-matched peers with normal cognition. In people with dementia, however, the marker returned to nearly normal levels. Soluble TREM2 levels directly correlated with elevated total tau and phospho-tau in the CSF of sporadic AD patients. Haass also saw elevated soluble TREM2 in people with normal cognition who carry a familial AD mutation, and in AD mouse models. It is not yet clear why, but one possibility is that high TREM2 represents an inflammatory defense against disease progression, Haass told Alzforum.
When TREM2 is shed, it leaves the cell surface. Many prior studies reported that TREM2 on microglia promotes phagocytosis (see Feb 2015 news; Hickman and El Khoury, 2014). Microglia without the receptor cannot devour Aβ in vitro, Haass noted. Likewise, microglia with T66M TREM2 poorly phagocytose beads, but when Haass blocked shedding of TREM2 by inhibiting ADAM10, phagocytosis improved. Haass believes faulty phagocytosis underlies disease, but stressed that the problem is not just removal of Aβ. Rather, dysfunctional microglia may fail to clean up cellular debris more broadly, intensifying neuronal stress and precipitating disease, he proposed. Other studies reported no difference in amyloid plaques in mice with little TREM2 (see Jun 2014 news).
Daniel Sevlever of the Mayo Clinic in Jacksonville, Florida, elucidated the effect of various TREM2 mutations. Sevlever reported that a mouse microglial cell line expressing the D87N mutation linked to AD shed less TREM2 into the medium than wild-type, and also had scant cell-surface expression of TREM2. On the other hand, cells with the R47H mutation shed TREM2 normally, agreeing with Haass’ data. The R47H mutation caused other problems, however, leading to reduced expression of TREM2 on the cell surface. Sevlever also looked at what happened to the soluble portion, which can interact with TREM2 ligands on neurons just as membrane-bound TREM2 does. He found that the soluble domain of R47H TREM2 bound poorly to the surface of neuroblastoma cells. Overall, this suggests that the R47H mutation may act through a different mechanism than T66M or D87N, perhaps by reducing binding or cell-surface expression rather than maturation and shedding.
Meanwhile, Karel Otero Gutiérrez of Biogen Idec, Boston, added data that further support TREM2’s role in brain cleanup. He fed mice cuprizone, a chemical that causes oligodendrocytes to die. In this model, the myelin sheath around axons falls apart, activating microglia to mop up the debris. In TREM2 knockout mice, microglia failed to activate or proliferate, and myelin chunks continued to litter the brain. These knockouts had worse motor problems than wild-type mice on cuprizone. Otero saw no signs of a pro-inflammatory effect in their brains, suggesting that defective activation and phagocytosis was the culprit (see Cantoni et al., 2015). “TREM2 may play a general role in controlling the activation of microglia in response to tissue injury,” Otero wrote to Alzforum. He noted that other work suggests TREM2 acts not just by facilitating phagocytosis, but also by supporting microglial survival in diseased and damaged brains.
Bucking the TREM2 tide, Jonathan Cherry, a graduate student with Kerry O’Banion and John Olschowka at the University of Rochester Medical Center, New York, argued that microglia expressing the marker arginase-1 are uniquely in charge of plaque removal. This enzyme was classically associated with M2 microglia, wound healing and suppression of nitric oxide production (see review by Cherry et al., 2014). Examining APPPS1 mice, Cherry found that in areas of chronic inflammation, arginase-1 expression soared while Aβ deposits plummeted. The expression of an anti-inflammatory marker in regions of high inflammation seemed counterintuitive to Cherry, and suggested microglia might be offsetting the inflammatory response. To study this, Cherry induced inflammation in mice. One month later, he found arginase-1 expression on microglia surrounding plaques. These cells had ingested twice the amount of plaque material as microglia expressing the traditional M1 marker iNos. Cherry confirmed that the arginase-1-positive cells were microglia, not macrophages, by using a chimeric mouse with fluorescently labeled bone marrow.
The finding suggested that arginase-1 positive cells might be more efficient phagocytes than their comrades. To confirm this, Cherry induced arginase expression with IL-4 injections; this led to a drop in plaques. Then he knocked out arginase-1 by infusing anti-IL4Rα through a minipump for 28 days. The treatment cut arginase-expressing cells in half and doubled the Aβ load, while not affecting cells positive for iNos. Together, Cherry concluded, his experiments demonstrate that arginase-positive microglia are necessary and sufficient for plaque removal, and validate arginase induction as a therapeutic approach.
Microglia may play a role in Parkinson’s disease, too. Dan Frenkel of Tel Aviv University suggested a mechanism by which the Parkinson’s risk genes DJ-1 and PINK1 could act on microglia to precipitate PD. He knocked down both DJ-1 and PINK1 in cultured microglia, and found the cells became more sensitive to activation, more neurotoxic, and expressed more pro-inflammatory markers (see Trudler et al., 2014). Moreover, when he stimulated the microglia with α-synuclein, he saw they gobbled up only about half as much as did microglia expressing those two genes. Curiously, the defect was specific to α-synuclein, as the cells phagocytosed yeast and inorganic particles normally. Frenkel noted that α-synuclein binds to lipid rafts on the cell membrane, structures that dwindle in cells lacking DJ-1 and PINK1. Deficient cells also had weaker autophagy, suggesting they might degrade α-synuclein poorly as well. Overall, the results hint that mutations in DJ-1 and PINK1 could alter microglia toward a neurotoxic phenotype, triggering disease and allowing α-synuclein deposits to accumulate, Frenkel proposed.—Madolyn Bowman Rogers
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C.
TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis.
Sci Transl Med. 2014 Jul 2;6(243):243ra86.
PubMed.

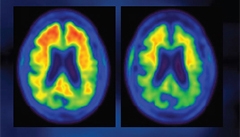



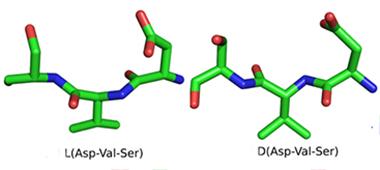







Comments
No Available Comments
Make a Comment
To make a comment you must login or register.