Learn the Skinny on iPS Cells in Neurodegenerative Disease
( 4 Articles Available, 0 Articles Pending )
Are you curious about trying iPS cell lines to model the disease you care about? Intrigued but nebulous on where the field is at? Ready to grow an iPS line but not sure where to turn? Read Madolyn Rogers's four-part series to learn all about who does what, which lines are going to be available when and where, and why and how they were made.
Rogers did not stop at the five Ws, though. She also reported the technical and scientific challenges this burgeoning field faces at present. Do those lines model Alzheimer's? Parkinson's? ALS? How pluripotent are iPS cells? Are the lines validated against an agreed-upon control? Read all, and be better off planning your research.
Image caption: Dopamine-producing neurons made from iPS cells derived from a Parkinson's patient. Image credit:: Frank Soldner and Dirk Hockemeyer/Whitehead Institute.
Where in the World Are the iPS Cells?
Since the discovery, in 2006, that adult cells could be reprogrammed to a pluripotent state, scientists have been excited by the potential of these cells for modeling diseases, screening drugs, and even for cell replacement therapy. The promise of induced pluripotent stem cells (iPSC) led Science magazine to name them as their Breakthrough of the Year in 2008 (see ARF related news story). Among other advantages, these adult-derived stem cells sidestep the ethical issues involved in embryonic stem cell (ESC) research, particularly relevant given renewed legal wrangling over federal funding for ESC experiments (see Dow Jones Newswires story). Nonetheless, scientists must still solve numerous technical challenges before the possibilities of iPS cells can be realized. Two years after Science’s celebration of iPS cells, it seems like an opportune time to survey the state of the field as it affects neurodegenerative disease research. What iPSC lines specific for these diseases are available to researchers now, or will soon become available? Who is making them? What are some of the technical challenges with these cells?
This reporter sought to answer these questions by interviewing 15 scientists in the field. However, this story can’t be comprehensive, and we invite our esteemed readers to fill in any omissions. One thing is clear: Neurodegenerative iPSC lines are mushrooming. Though only a handful of lines have been published to date, a plethora of lines are scheduled to appear in print or be accessible from cell banks within the next few months, for diseases such as Alzheimer’s, Parkinson’s, Huntington’s, ALS, and frontotemporal dementias. Despite the rush to make lines, the field has not yet reached a consensus on the best way to generate these cells, on the best way to differentiate them into mature cell types, or on what is required to validate these cell lines as truly pluripotent and useful. Moreover, iPSC lines harbor tremendous genetic and epigenetic variation, which may pose a problem for comparing data between experiments. (See Part 4 of this series for a discussion of these issues.)
The iPS Cells Are Coming: A Snapshot of Summer 2010
While there are no standardized protocols yet, three common themes are emerging among scientists who generate iPSC lines. For one, most labs continue to use retroviral or lentiviral reprogramming due to the efficiency and ease of this method, rather than using newer non-integrating methods. For another, many researchers are making not only their iPSC lines publicly available, typically through a cell bank, but also the original fibroblast lines they used to generate the iPSCs. This allows other labs to employ their own method of choice to reprogram fibroblasts carrying mutations of interest. Finally, in addition to private labs working on iPSC lines, disease foundations, cell banks, and large consortia are also getting involved. For more on each of these points, and details on disease-specific iPSC lines, see Part 2, Part 3, and Part 4 of this series.—Madolyn Bowman Rogers.
In Alzheimer Disease Research, iPS Cells Catch On Slowly
While scientists are steaming ahead on establishing iPSC lines for research into Parkinson disease and amyotrophic lateral sclerosis (ALS) (see story below and Part 3), they have been more reluctant to embrace the technology for Alzheimer disease. Perhaps this is due to concerns that a complex, late-onset disease such as AD cannot be easily modeled by iPS cells. Nonetheless, a few labs are creating iPSC lines from dementia patients. Selina Wray, working with John Hardy and Patrick Lewis at University College London, U.K., is making multiple iPSC lines specific for various familial dementias, including AD and frontotemporal dementia. Patients in the cognitive disorders clinics at UCL donate the necessary fibroblasts, and because the patients have been genotyped at the clinic, their mutations are known. Many of the clinic patients are also part of the Dominantly Inherited Alzheimer Network (DIAN, see also ARF conference story), Wray said. This means they have brain images and fluid samples on file as well, providing detailed clinical information to correlate with data from iPSC lines. Hardy’s group also collects fibroblasts from relatives of the patients who carry the same mutation, as well as from unaffected relatives for use as controls. Wray added that she is using the original 2006 Yamanaka method (see Takahashi et al., 2006) and retroviral vectors to reprogram the fibroblasts, because of this method’s efficiency. The London group will make both their iPSC and fibroblast lines available and is looking at depositing them either with Coriell Institute for Medical Research in Camden, New Jersey, or with the European Collection of Cell Cultures (ECACC). The fibroblasts might be available within six months, Wray said, but she expects the iPSC lines to take longer due to the need to validate each line.
Wray currently has several iPSC lines with tau mutations, which cause frontotemporal dementia, as well as lines for one APP mutation and one presenilin mutation, both of which cause Alzheimer’s. She said that she expects to generate many more lines, with the goal to get at least three lines of each mutation for comparison. Wray does not know yet if neurons generated from these lines will show disease traits in culture, but she is excited by the potential of iPS cells. “I think they really will open a huge range of possibilities and give us a much more physiologically relevant system, because we’ll be looking at the mutated proteins at endogenous levels,” Wray said. She believes iPS cells will provide a better model for studying tau splicing, for example, than do either current animal models or knockdown and overexpression experiments. “The pattern of tau splicing is specific to humans,” Wray said. There is debate in the field about splice forms of tau in mouse and rat models, raising the question to what extent rodent models reflect the human disease (see ARF ICAD news story).
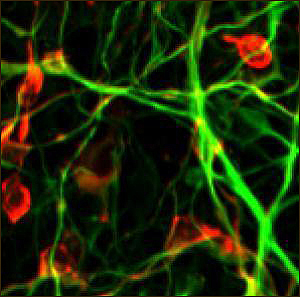
Dopamine-producing neurons made from induced pluripotent cells, reprogrammed from the skin of a Parkinson's patient. Green is a neuronal marker (class III β-tubulin) and red is a dopamine-producing enzyme (tyrosine hydroxylase). Image credit: Frank Soldner and Dirk Hockemeyer/Whitehead Institute
Other researchers making iPS cells from AD patients include Larry Goldstein at the University of California in San Diego, and Asa Abeliovich at Columbia, New York City. Goldstein said his lab is generating iPSC lines from fibroblasts of people with APP duplications and presenilin mutations, as well as from a few patients with sporadic AD. They have already differentiated these iPS cells into neurons and characterized them, and Goldstein expects the lines will be available from his lab upon request within a few months. Goldstein’s work is supported by the National Institute on Aging (NIA), using funds from the American Reinvestment and Recovery Act. Abeliovich is working on both AD and PD iPSC lines. Abeliovich said he is generating lines from both sporadic and familial forms of the diseases, but they are not yet available.
Parkinson Disease
The second most common neurodegenerative disease, Parkinson’s, has been an early focus of iPSC research, with several labs and large groups creating PD-specific lines. Among the handful of published iPSC lines are some early ones generated from five patients with sporadic PD by researchers led by Rudolf Jaenisch of MIT (see Soldner et al., 2009). These lines are available now, Jaenisch said; however, they are not useful as disease models because their genetics are unknown, and they have so far failed to show a disease phenotype in vitro. “The key now is to generate lines that are genetically defined,” Jaenisch said. His group is developing iPSC lines from PD patients with known mutations, using a Cre-Lox system that removes reprogramming factors (see ARF related news story on Soldner et al., 2009).
Working with Hardy at University College, Lewis and Helene Plun-Favreau are also making iPSC lines from familial PD patients. They are currently generating a synuclein triplication line and about a dozen lines with mutations in LRRK2, said colleague Wray. When finished, these lines will be made publicly available through a cell bank.
Here’s yet another group active in this field. While in George Daley’s lab at Harvard, first author In-Hyun Park, now at Yale in New Haven, Connecticut, created one iPS line each from people with PD, Huntington disease, and Down syndrome (see Park et al., 2008). These lines are all available through the iPS Core of the Harvard Stem Cell Institute. Park said he created these lines as a proof of concept of the versatility of iPS cells, but is not continuing to pursue them as disease models.
Large foundations and multi-institution consortia are also diving into the iPSC field. The nonprofit Parkinson’s Institute and Clinical Center in Sunnyvale, California, is working with the Stanford Institute for Stem Cell Biology and Regenerative Medicine and the Department of Neurosurgery at Stanford to develop iPSC lines from patients with both sporadic and familial forms of PD, according to information on the Parkinson’s Institute website. The project is funded by the California Institute for Regenerative Medicine, and cell lines will be available to researchers within California, wrote Birgitt Schuele at the institute in an e-mail to ARF. The institute plans to establish a bank of 15-20 iPSC lines, differentiate the cells into dopaminergic neurons, and use them to model the disease and screen drugs. So far, they have generated the first iPSC lines from patients with mutations in the SNCA gene that encodes α-synuclein.
The Role of Consortia One of the largest multi-institution iPSC efforts draws support from the National Institute of Neurological Disorders and Stroke (NINDS). With funds from the American Reinvestment and Recovery Act, NINDS is supporting three consortia with the goal of creating iPSC lines from familial forms of HD, PD, and ALS, wrote Margaret Sutherland, program director at NINDS, in an e-mail to ARF. Each consortium will contribute fibroblasts and iPSC lines to the NINDS Repository at Coriell. The lines will be provided to academic and not-for-profit investigators at cost. Sutherland said that one of the consortia’s goals is to generate well-characterized cell lines, including profiles of gene expression and epigenetics (see ARF related news story on NIA epigenetics research). Some of the lines may include cell-specific reporters to allow scientists to monitor the differentiation state of the cells. The consortia have also been charged with developing criteria to define high-quality iPS cell lines, Sutherland said. Each consortium is working on a two-year time frame that will be completed 1 October 2011. To date, each consortium has deposited about 10 to 15 cell lines with Coriell, Sutherland said, and these lines may become available to academic and industry scientists in early 2011.
The HD consortium is led by Leslie Thompson at the University of California, Irvine, and includes co-principal investigators Steve Finkbeiner, Clive Svendsen, Chris Ross, Marcy MacDonald, and Jim Gusella, at institutions around the country. The group will look at a series of increasing CAG repeat lengths and examine the associated phenotypes, Sutherland said. The PD consortium is under the charge of Ole Isacson at Harvard and includes co-PIs Virginia Lee, John Trojanowski, Ted Dawson, Jim Surmeier, and Karen Marder, from several institutions. See Part 3 of this series for a discussion of the ALS consortium, and neurodevelopmental iPSC lines.—Madolyn Bowman Rogers.
Hereditary Diseases: A Natural Fit For iPSC Modeling
One of the three iPSC consortia funded by the National Institute of Neurological Disorders and Stroke (NINDS) is creating lines for amyotrophic lateral sclerosis (ALS). The ALS consortium is headed by Jeff Rothstein at the Robert Packard Center for ALS Research at Johns Hopkins University, Baltimore, Maryland. The scientists are developing iPSC lines for 25 familial forms of ALS, Rothstein said, as well as control lines from healthy people. The Johns Hopkins team will differentiate the iPS cells into astrocytes, while co-PI Chris Henderson at Columbia University, New York, will generate motor neurons from the cells. Two other co-PIs, Tom Maniatis at Columbia and Kevin Eggan at Harvard, will handle iPS cell profiling and biology, Rothstein said.
One of the potential applications of iPSC-derived cells carrying ALS mutations is for drug discovery. However, “We don’t know yet if [iPSC-derived cells] will be better than using a rat,” Rothstein said. Because it’s faster to use neurons from existing transgenic rat and mouse ALS models than to generate iPS cell lines from humans, Rothstein predicted that rodent cells might remain the method of choice for initial drug screening, with iPSC-derived cells serving a second step of verifying promising drug candidates. The Johns Hopkins team has also generated about 20 lines from sporadic ALS patients, Rothstein said, but these lines are not yet useful as disease models. Rothstein said that is because sporadic ALS cells contain too many unknowns; scientists will need to characterize cells with known mutations before they can make sense of lines from sporadic cases.
For his part, Eggan said that besides his work with the consortia, he also generates his own ALS iPSC lines and has already published several (see ARF related news story on Dimos et al., 2008 and ARF related news story). These lines will be available to other research groups through the iPS Core of the Harvard Stem Cell Institute (HSCI), and many will go online within a few weeks, Eggan said. To date, Eggan and colleagues have made multiple ALS lines from about a dozen patients, including various TDP-43 mutations and an allelic series of SOD1 mutations. His group has also generated control lines from both relatives of patients and unrelated folks. The scientists are using retroviral programming with three factors, a method that Eggan said is almost 100 percent successful in producing iPSC lines from every patient. The lab is currently trying to identify a phenotype in cell cultures made from these iPSC lines, Eggan said. In addition, his lab is working on iPSC lines for the inherited degenerative disease spinal muscular atrophy (SMA), but these lines are not yet ready to distribute.
Neurodevelopmental and Single-Gene Diseases—Voila, a Phenotype!
Monogenetic diseases, such as SMA, are obvious candidates for modeling with iPS cells, because the genetics are simple and the diseases usually manifest earlier. The SMA iPSC line made by first author Allison Ebert, working in Clive Svendsen’s lab at the University of Wisconsin in Madison, has the distinction of being one of the few published iPSC lines to demonstrate a disease phenotype in culture (see Ebert et al., 2008). The published iPS lines were made using a lentiviral vector and reprogramming factors identified by James Thomson at the University of Wisconsin (see Yu et al., 2007). Both the iPS cells and the fibroblasts are available through Coriell Institute for Medical Research in Camden, New Jersey, said Ebert’s colleague Jered McGivern. The lab is currently making additional SMA lines for comparison, McGivern said. The scientists are generating some of the new lines using a non-integrating episomal vector, also developed by Thomson (see Yu et al., 2009).
Induced pluripotent stem cells are particularly valuable for studying SMA, McGivern said, because the disease only occurs in humans. “There are some model systems in mice, but they may not recapitulate what happens in humans,” he added. McGivern said the lab also has some Huntington disease-specific iPSC lines that were made with fibroblasts from Coriell and reprogrammed in the Thomson lab.
Other published iPSC lines for monogenetic and neurodevelopmental diseases include a fragile X iPSC line created by researchers working with George Daley at Harvard (see Urbach et al., 2010). There is also an iPSC line for familial dysautonomia—an inherited disorder in which autonomic and sensory nerves malfunction—made by researchers led by Lorenz Studer at Sloan-Kettering Institute in New York (see Lee et al., 2009). The familial dysautonomia line has shown a disease phenotype in culture. James Ellis’s group at the University of Toronto, Canada, published an iPSC line from a Rett syndrome patient (see Hotta et al., 2009). Ellis said he is currently collecting biopsies from patients in his pediatric clinic, and using them to generate more iPSC lines for neurodevelopmental disorders such as Rett syndrome, autism, and schizophrenia, as well as making control lines from relatives of patients. These lines will initially be available from Ellis, later through the Ontario Human Induced Pluripotent Stem Cell Facility.
Control Lines
To use all of these disease-specific iPSC lines for disease modeling, researchers will also need control lines from healthy people. Most research groups are making their own control lines from relatives of patients, but some lines are also commercially available, or will be soon. The stem cell bank at the University of Wisconsin in Madison, WiCell, stocks several iPSC lines made by Thomson using both lentiviral and episomal vectors.
Coriell, a nonprofit biomedical research institution, not only banks iPSC lines from many other labs, but is also developing its own lines. At the moment, Coriell is focusing on making iPSC lines from fibroblasts taken from healthy people, Margaret Keller, director of Coriell’s stem cell bank, wrote to ARF; future plans include making iPSC lines from people with diseases. This work, which is sponsored by the National Institute of General Medical Sciences (NIGMS), currently uses a lentiviral reprogramming protocol based on the one developed by Thomson, Keller said, though they may also use a non-viral method in the future. Coriell will make both the iPSC lines and fibroblasts available to researchers, with the first lines predicted to come online in late 2010, Keller said. Coriell also banks numerous lines from other researchers, many of which are expected to become available through the NIGMS Repository collection in late 2010.
Clearly, there will soon be numerous cell lines available for researchers interested in studying neurodegenerative diseases with iPS cells. This brings to the fore a question that has been lurking just beneath all this effort. How useful are these lines as disease models? What limitations should researchers keep in mind? For an answer to these questions, read Part 4 of this series.—Madolyn Bowman Rogers.
Since the 2006 discovery by Shinya Yamanaka and Kazutoshi Takahashi at Kyoto University, Japan, that just four transcription factors could reprogram mouse fibroblasts to a pluripotent state, induced pluripotent stem cells have created excitement among researchers who see them as more available, easier to make, and less ethically conflicted than embryonic stem cells (ESCs). Generating several iPSC lines takes about six months, compared with 18 months to obtain one ESC line, said Mahendra Rao, vice president of stem cells at Life Technologies, a biotechnology tools company. “It’s really changed the embryonic stem cell field,” Rao said. “It’s made pluripotent cells available to people who don’t have to be experts.”
Reprogramming Riddles
Yet scientists in this field still face numerous challenges. One concern, which has been covered extensively on ARF, is the use of retroviral or lentiviral reprogramming vectors, which integrate into the genome. Most researchers contacted for this story feel that iPS cells reprogrammed with integrating viruses are unsuitable for cell replacement therapy, but are acceptable for cellular disease models. Nonetheless, researchers led by Rudolf Jaenisch at MIT found that residual expression of reprogramming genes in iPS cells may interfere with fully resetting the cells to an embryonic pluripotent state. When Jaenisch and colleagues removed the reprogramming genes using a Cre/Lox system, the iPS cells’ gene expression profile matched up better to that of embryonic stem cells than did iPS cells that contained reprogramming genes (see Soldner et al., 2009). This suggests that removing reprogramming factors may be important even for disease modeling. Researchers have now demonstrated the efficacy of numerous next-generation reprogramming methods that leave no trace of themselves in the cell (see ARF related news story on Stadtfeld et al., 2008; ARF related news story on Okita et al., 2008; ARF related news story on Kaji et al., 2009, Soldner et al., 2009, and Woltjen et al., 2009; and ARF related news story on Yu et al., 2009). The problem with all of these methods is that the already low reprogramming efficiency drops further, usually by a couple of orders of magnitude, causing most scientists interviewed here to continue to opt for the higher-efficiency viral methods for the time being.
Scientists also stew over the quandary of how thoroughly cells truly are reset to pluripotency. Some new research suggests that iPS cells may not be fully reprogrammed to a stem cell-like state. For example, research groups led by George Daley and Konrad Hochedlinger, both at Harvard, independently found that iPS cells can contain an epigenetic “memory” of their tissue of origin, which restricts their differentiation potential (see Polo et al., 2010 and Kim et al., 2010). Hochedlinger’s group found that continuous passaging of the pluripotent cells caused this memory to fade, while Daley’s group was able to reset these cells using serial reprogramming or chromatin-modifying drugs. On the other hand, new work by Jaenisch’s group published last month in Cell Stem Cell analyzed gene expression and epigenetic profiles of iPSCs and ESCs and concluded that there are no significant, consistent differences (see Guenther et al., 2010). This report engendered some debate, as William Lowry and colleagues at the University of California, Los Angeles, contend that some iPSC-ESC differences seen by various labs are indeed significant (see Chin et al., 2010). Lowry’s group concurs, however, that variables such as delivery of the reprogramming factors on a single vector, and high passage number (50-80 passages), reduce the observed differences between iPSCs and ESCs.
Many scientists interviewed for this story agreed that it is important to be aware of the issue of incomplete iPSC reprogramming. At the same time, few consider it a disabling problem, as it can be solved by longer passage times or better reprogramming methodologies. If the differentiation potential is restricted, that is not necessarily a bad thing, said In-Hyun Park at Yale in New Haven, Connecticut. Scientists can use cells of the desired lineage to generate iPSCs, for example, neuronal lineage cells to generate iPSCs for neuronal research. Kevin Eggan of Harvard suggested that for disease models, the question of whether iPSCs and ESCs are exactly the same might be less important than whether iPSCs can make the functional cell types of interest and whether those cells show a phenotype in a culture dish.
Recapitulating Disease in a Dish
Indeed, the latter ability is one of the crucial traits that iPS cells must possess if they are to realize their potential as disease models. So far, very few published lines have demonstrated disease features in culture. Two notable examples that did show disease traits both modeled early onset, monogenetic disorders, that is, spinal muscular atrophy and familial dysautonomia. Many researchers interviewed for this series voiced concern that sporadic forms of disease may simply not show up in iPS cells, in part because these disorders may result from environmental factors that change the epigenetic profile of the neurons. iPS cells, which have a nearly fresh epigenetic slate and are often derived from a different tissue to boot, would not mimic these disorders.
Scientists also remain skeptical at this point about whether late-onset diseases such as AD will show defects in a cell culture. “I think the concern in the field for Alzheimer’s cells is that you would have to culture them for an enormous amount of time,” said Jered McGivern, University of Wisconsin, Madison, who works with Clive Svendsen, now at Cedars-Sinai Medical Center, Los Angeles, California. Some solutions have been proposed, such as genetically modifying AD cell lines to stress or age them more quickly, or using human-animal chimeras (see Saha et al., 2009), but these suggestions still await testing. Disease modeling with iPS cells remains a frontier that scientists have only just begun to explore.
Here is another sticky issue for modeling. Can a cell culture that contains but one or two cell types ever successfully represent a complex disease? Some of these diseases, such as schizophrenia, many forms of epilepsy, and perhaps AD, are probably circuitry disorders, said Rao. “Anything you do on a dish in 2D is not going to reconstruct the normal neuronal circuitry you’d see in a three-dimensional brain,” concurred James Ellis, of the University of Toronto in Canada. Disease modeling remains problematic for another reason, Ellis said. That is, scientists lack efficient differentiation procedures for creating adult-phenotype, subtype-specific cells of interest without also getting undesired cell types.
Vexing Variability
The variability of iPS lines presents one of the biggest obstacles for scientists hoping to use these lines for research, Park said. Because of natural genetic variation between one person and the next, iPS lines from different patients are hard to compare. Park said he also sees differences even among iPS cells derived from the same fibroblast line, indicating that some of the variation is due to the reprogramming process. In addition, Ellis pointed to new research showing that the expression profiles of iPSCs and ESCs are affected by the laboratory that makes them (see Newman and Cooper, 2010), suggesting that culture conditions significantly alter gene expression. All these factors may make iPS data too noisy to recognize a phenotypic difference between a single mutant line and a single wild-type line, Park said, adding, “It’s going to be difficult to generate an in vitro model of a disease.”
Asa Abeliovich, of Columbia University in New York City, agreed that this variability is a key concern with using iPS cells to model disease. Abeliovich points to the importance of comparing multiple iPSC-derived lines and using statistical analysis to determine the ratio of signal to noise in the system. Without such analysis, Abeliovich said, you can’t be sure that a phenotype is real. Abeliovich emphasized this point while presenting initial data on his lab’s iPS cell lines at the International Conference on Alzheimer’s Disease in July 2010 in Honolulu, Hawai’i.
“We think what’s going to happen is that researchers will want their own lines, from their specific patient set,” Rao said, adding that particularly for complex diseases such as AD, having a clinical history to correlate with the iPSC data will be important. The iPSC line variability means that having proper control lines is critical, but there’s no consensus in the field yet as to what lines will make the best controls. Many institutions try to gather control lines from a patient’s relatives, to minimize the effect of the genetic background. For monogenetic diseases, Jaenisch suggested in a paper, the most stringent control might be to genetically rescue some iPS cells, so that the only variation between rescued and mutant cells is the gene of interest (see Saha et al., 2009). iPSC variability will also make it harder for labs to compare data. Rao said it will be important for the science community to have a commonly used, standard reference line. Adding a plug, he noted that Life Technologies plans to develop such a reference iPS line.
Despite all these hurdles, most researchers remain optimistic about the potential of iPS cells to make useful disease models and research tools. “I think the opportunities that [iPS cells] present outweigh the limitations,” said Selina Wray, of University College London, U.K. Rao agreed, saying, “Depending on the question you’re asking, some of these problems are not relevant. We’ve always lived with the fact that we can never have a perfect model.” Induced pluripotent stem cells will be particularly valuable for disorders such as motor neuron diseases, Rao said. Because motor neurons can’t be easily cultured or genetically manipulated, scientists have never had a good model system for some of these diseases. “iPSC now offers you a way to do that,” Rao said. “Stem cells have the promise of providing us entry into a black box of development that we simply had no other way to look at.”—Madolyn Bowman Rogers.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.