CONFERENCE COVERAGE SERIES
Alzheimer's Association International Conference 2013
Boston, MA, U.S.A.
13 – 18 July 2013
CONFERENCE COVERAGE SERIES
Boston, MA, U.S.A.
13 – 18 July 2013
Despite rampant plaque pathology, most transgenic mice that overproduce amyloidβ (Aβ) fail to capture other hallmarks of Alzheimer’s disease, such as neurofibrillary tangles and neuronal loss. Now, a study in the July 17 Science Translational Medicine suggests that some of these models may be better than previously thought. Scientists led by Mathias Jucker, University of Tübingen, Germany, report that changes in mouse cerebrospinal fluid (CSF) tau and Aβ look strikingly similar to those of people with sporadic or familial Alzheimer’s disease. These animals could help predict how human CSF Aβ and tau may shift in response to drugs in early-stage trials, and may help researchers develop new CSF-based markers, the authors wrote.
“The study suggests that animal models of amyloidosis can help us model aspects of human AD that we didn’t think they could before,” said David Holtzman, Washington University, St. Louis, Missouri.
First author Luis Maia presented this data at the Alzheimer's Association International Conference, which ran July 13-18 in Boston, Massachusetts. His presentation came hot on the heels of a well-received plenary talk by David Borchelt, University of Florida, Gainesville, entitled "Are our animal models good enough?" Borchelt noted that while potential AD therapeutics look impressive in various mouse models, none have translated into an approved treatment. He contended that since their cognitive deficits are subtle and they retain the vast majority of their synapses and neurons, many of these mice model prodromal Alzheimer's rather than symptomatic. Maia agreed in his talk, suggesting that the emerging biomarker data for APP/PS1 and APP23 mice fits the prodromal profile. Other researchers at the meeting challenged that view. For example, Kelly Dineley, University of Texas Medical Branch, Galveston, suggested that the cognitive decline in the mice was more akin to symptomatic AD.
Previous studies reported that CSF monomeric Aβ levels fall as transgenic mice age and develop Aβ plaques (see Kawarabayashi et al., 2001 and Liu et al., 2004). That recapitulates the pattern seen in people who develop AD. As more Aβ deposits in the brain, less of the soluble peptide winds its way into the CSF. However, few researchers thought to look for CSF tau changes, Jucker told Alzforum. This was partly because tau was not expected to show up in CSF since the animals have no neurofibrillary tangles and little neurodegeneration, and partly because the teeny volume of mouse CSF (about 5-10 uL) and the low concentration of tau made the protein difficult to measure. Maia and co-first author Stephan Kaeser used a highly sensitive antibody-based chemiluminscent assay developed by the company Meso Scale Discovery to detect both tau and Aβ.
The researchers used a method previously developed in Holtzman’s lab to almost double the yield of mouse CSF. It keeps anaesthetized animals alive long enough to drain the fluid twice. Since the protocol requires animal sacrifice, Maia and colleagues sampled five different groups of mice at different time points over their lifespan. In the APPPS1 mice, Aβ42 fell by half at six months of age and by 80 percent at 18 months. Aβ40 dropped, too, but to a lesser extent: only 45 percent by 18 months. In the APP23 mice, Aβ42 decline began around 16 months, and reached a 60 percent drop by 30 months. In both of these cases, CSF Aβ levels inversely correlated with rising Aβ deposits in the brain as seen by immunocytochemistry and Congo red staining.
To the researchers’ surprise, endogenous mouse tau rose in the CSF of both models shortly after Aβ levels began to drop. For APP/PS1 mice this was at six months, with tau surging to five times above baseline level by 18 months. The upshot was a 45-fold increase in the tau:Aβ42 ratio. In the APP23 mice, tau also rose about five-fold relative to baseline at 24 months, amounting to a 10-fold hike in the tau:Aβ42 ratio. In both types of mice, the rise occurred in the absence of global neuron loss, neurofibrillary tangle formation, or change in total tau in the brain. Many neuritic structures surrounding Aβ deposits contained phosphorylated tau, however. Meanwhile, levels of α–synuclein stayed the same, suggesting no overall release of cytoplasmic proteins from neurons.
These results challenge current assumptions about the cause of rising tau in human CSF, wrote the authors. If it is unrelated to neuron loss or neurofibrillary tangle formation, why does tau end up in the CSF? The authors suggested that neurons might release tau during normal physiological activity (see Pooler et al., 2013), which is stimulated by Aβ. Alternatively, tau could leak from neurites in response to Aβ–induced damage, suggested Jucker. Several other scientists voiced support for the latter hypothesis. Holtzman in particular said he has suspected such a tau-release mechanism for a while, as CSF tau levels tend to be stable in people with non-Aβ-related tauopathies (see Hall et al., 2012 ). If Aβ causes tau release, then anti-Aβ therapies might reduce CSF tau. On the other hand, tau therapies might not lower CSF tau because Aβ would continue to promote its release into the fluid.
CSF profiles in these mice do not exactly match those of humans with mild cognitive impairment or AD, noted Douglas Galasko, University of California, San Diego, in an email to Alzforum (see full comment below). He pointed out that CSF Aβ42 levels fall lower and tau levels rise higher in these mice, and that CSF Aβ40 does not typically fall in humans. “Nevertheless, the time course of these changes is highly relevant to evaluating the impact of treatment interventions, with the hope that predictions used from brain and CSF analysis in mice could help to guide translation into human studies,” he wrote.
The results give the first real clinical significance to these mouse models, Jucker said, because they can help foretell how drugs will affect key CSF biomarkers in people, especially those with the particular familial AD mutations carried by the mice. He plans to observe how CSF Aβ and tau levels change in these models in the presence of BACE inhibitors, γ-secretase inhibitors, and various immunizations. He also plans to develop an assay to detect phosphorylated tau, which his group was unable to measure in this study.
Longitudinal mouse studies would be highly informative. They would possible if researchers developed assays that require less CSF so that small volumes could be drawn from mice without being lethal, said Henrik Zetterberg, University of Gothenburg, Sweden. Zetterberg called for further research to figure out how amyloid stimulates the release of tau in these models.—Gwyneth Dickey Zakaib and Tom Fagan
No Available Further Reading
Insurers have spoken: brain amyloid β PET imaging is not ready for clinical prime time. On July 3, the Centers for Medicare and Medicaid Services (CMS) issued a draft decision denying coverage for any clinical use of the technology. The decision met with strong criticism at the Alzheimer's Association International Conference (AAIC), which ran July 13-18 in Boston, Massachusetts. At a special public policy session held July 15, Alzheimer’s disease (AD) stakeholders lambasted the decision and strategized possible responses. Led by the association’s Maria Carrillo, this gathering saw some of the conference’s most passionate debate.
The session attracted a packed room of academic researchers, clinicians, and representatives from industry, imaging associations, and advocacy groups. Across the board, these stakeholders expressed frustration and disappointment with the CMS decision, decrying the loss of what they unanimously see as a valuable tool for patient care. The silver lining is that CMS will reimburse for scans in clinical trials that meet specific criteria; however, the overall decision will greatly limit access to amyloid scans in clinical care. An amyloid scan can cost $3,000 or more. Not only are most Alzheimer’s patients Medicare beneficiaries, but private insurers tend to follow CMS’ lead and are expected to deny reimbursement for this procedure, as well. “Having our hands cuffed behind us as we try to help patients and their families is a moral dilemma,” said Michael Devous of the University of Texas Southwestern Medical Center, Dallas.
The group also voiced their determination to take action. “This draft decision is a mistake and we should oppose it,” said Keith Johnson at Massachusetts General Hospital, Boston.
No insurance representatives spoke at the July 15 meeting. Instead, insurers aired their views at a separate July 18 AAIC session led by Steven Pearson, president of the Institute for Clinical and Economic Review (ICER). Based at MGH’s Institute for Technology Assessment, ICER, which receives unrestricted funds from several insurance companies, assesses the value of healthcare interventions across different medical fields. Pearson presented the findings from ICER’s white paper on the evidence standards needed for insurance coverage of AD diagnostic tests (see ARF related news story). Insurers will only cover tests that improve patient health outcomes, Pearson emphasized. A panel that included several insurance representatives agreed with this standard and discussed what evidence might be needed for amyloid scans. This session generated little crosstalk with AD researchers, as the audience was sparse and the panel took but a single question as time expired.
Insurers and Clinicians Clash Over Benefits of Amyloid Imaging
Amyloid β imaging became clinically available in 2012, when the Food and Drug Administration approved the first commercial Aβ PET ligand, Eli Lilly and Company’s Amyvid (see ARF related news story). To help guide its clinical use, the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging convened a taskforce to develop appropriate criteria, which were published in January this year (see ARF related news story). AD stakeholders of every stripe have embraced the criteria. “I’ve never seen a coalition of common thought like we have in this setting,” noted Devous at the July 15 meeting.
In December 2012, Lilly petitioned CMS to extend coverage for amyloid β PET imaging agents (see ARF related news story). In response, CMS opened a National Coverage Analysis, which is the agency’s routine procedure for reconsidering coverage policies. In January 2013, CMS convened a meeting of the Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to examine the issue. MEDCAC voted that there was not enough evidence to determine if amyloid scans improved patient health outcomes (see ARF related news story), presaging the negative draft decision.
The sessions at AAIC highlighted the gulf between insurers’ and clinicians’ points of view. “The value of a diagnostic test depends on the availability of effective therapies,” said Robert McDonough, who heads clinical policy at Aetna, at the July 18 AAIC session. The MEDCAC panel had expressed similar views, noting that in the absence of a disease-modifying AD therapy, correct diagnosis has little effect on patient health.
Clinicians strongly disagree. “Having an accurate diagnosis is the foundation of good medical care,” said Stephen Salloway of Butler Hospital, Providence, Rhode Island. He pointed out that diagnosis affects prognosis, medications, and patient education. Ron Petersen of the Mayo Clinic in Rochester, Minnesota, said that amyloid scans would be useful to rule in AD when people have atypical symptoms. Speaking at the July 18 meeting, Ali Shamsi at Yale University, New Haven, Connecticut, stressed the value of negative amyloid PET scans for ruling out AD when people present clinically with dementia that is due to a silent stroke, other brain injury, drug abuse, or chronic mental illness. In these cases, correct diagnosis would greatly alter treatment and decisions about where best to treat and care for patients, Shamsi said. In this way, amyloid imaging also aids differential diagnosis for people who have a non-AD progressive neurodegenerative disease, such as frontotemporal dementias. Norman Foster of the University of Utah, Salt Lake City, said, “We’ve been surprised that so many people coming to memory clinics have negative scans.” Many of the speakers at the July 15 session have served as consultants for Lilly.
CMS’ Answer: Coverage with Evidence Development
Now about that silver lining. In the draft decision, CMS agreed that PET Aβ imaging could be promising in specific scenarios, such as excluding AD in clinically difficult diagnoses, or to enrich clinical trials. To prove that scans improve patient outcomes in these scenarios, CMS proposed a “coverage with evidence development” (CED) process. The agency will cover a single PET Aβ scan for cognitively impaired Medicare beneficiaries participating in randomized clinical trials. These trials must address the question of whether amyloid imaging leads to improved health outcomes. They must also satisfy various other strictures, including “postmortem diagnosis as the endpoint where appropriate.”
Speakers at the July 15 session roundly condemned this last requirement as absurd, noting that an autopsy endpoint could require trials to last 15 to 20 years. Even without that criterion, AD stakeholders criticized the CED process as unnecessarily slow and burdensome. For example, one upcoming trial that is generating considerable excitement in the field, the Anti-Amyloid Treatment in Asymptomatic AD (A4) Trial, will use amyloid scans to screen presymptomatic patients for inclusion; these people are expected to live for decades.
Moreover, AD researchers wondered how such trials could be run in practice. “I’m concerned about how we would actually generate evidence of benefit,” said Reisa Sperling of Brigham and Women’s Hospital, Boston. She wondered what outcome metrics would be acceptable. Diagnostic tests by themselves do not improve symptoms or survival, Sperling noted. Even if paired with an experimental AD drug, it might be hard to see clinical change in a prodromal AD population. Would CMS consider improvement on cognitive tests to be a meaningful clinical outcome? Sperling asked. Researchers note that none of that is clear.
Previously, researchers in the field of oncology used the CED mechanism to gain coverage for PET ligands through the National Oncologic PET Registry (NOPR). This process took seven years. However, the bar has been set higher for Aβ ligands, said Gail Rodriguez, president of the Medical Imaging and Technology Alliance (MITA), a Washington, D.C.-based industry association. Under NOPR, PET scans were reimbursed for all Medicare beneficiaries as long as their physician submitted data regarding the treatment decision to the registry. Clinical trials were not required.
Numerous scientists perceived a double standard between coverage decisions on fluorodeoxyglucose (FDG) PET and amyloid PET. CMS covers the use of FDG PET to distinguish between frontotemporal dementia (FTD) and AD, but not amyloid PET, despite evidence that the latter is more effective for this purpose.
Some believe the real issues are financial. Rodriguez suggested that CMS wants to throttle access to the scans because it fears a deluge of demand for which it cannot afford to pay. Howard Fillit, director of the Alzheimer’s Drug Discovery Foundation, New York City, struck a moderate tone, noting that in the current climate of health care reform and cost-cutting, CMS’ focus on improved outcomes is reasonable. Insurers fear that everyone over 50 will want this test, Fillit said. He suggested reassuring CMS that primary care physicians will not be able to order the scans. In a plenary session on amyloid imaging, Stephen DeKosky at the University of Virginia, Charlottesville, noted that insurers are increasingly denying even FDG PET scans.
AD Stakeholders Weigh Options
At the July 15 meeting, participants strategized about ways to persuade CMS to change its ruling, and do so during the ongoing 30 day period in which CMS will accept public comments. Many advocated for flexing political muscle, pointing out that the CMS verdict clashes with the National Alzheimer’s Project Act’s (NAPA) directive to “optimize care quality” for AD patients (see ARF related news story; ARF related news story). Others suggested mobilizing a grassroots effort of Alzheimer’s Association members, Medicare recipients, general care practitioners, and neurologists. Researchers fretted over the limited time available before the draft decision is made final on August 2. Carrillo said the association will request that CMS extend the comment period to 90 days.
Many agreed that the best strategy is to press for coverage for certain limited indications, particularly where there is already a precedent for CMS coverage of FDG PET scans. In addition to the differential diagnosis of FTD versus AD, a precedent exists for coverage of early onset AD, said Jason Karlawish at the University of Pennsylvania, Philadelphia. Researchers agreed this might serve as a foot in the door toward eventual broader coverage.
Meanwhile, Lilly continues to gather evidence to try to support the clinical utility of amyloid β PET scans. At AAIC, Noam Kirson of The Analysis Group Inc., Boston, presented cost data from a study funded by Lilly. He reported that Medicare beneficiaries with Parkinson’s disease or vascular dementia who were incorrectly diagnosed with AD accumulated about $12,000 more per year in reimbursed Medicare expenses compared to peers who were correctly diagnosed. Once the misdiagnosis was corrected, their costs dropped back to those of peers. This would suggest that ruling out AD early could save money, Kirson concluded.
Another talk at AAIC correlated early diagnosis with better patient outcomes. Ilona Hallikainen from the University of Eastern Finland, Kuopio, reported that people with AD managed their daily activities better and had less psychological and behavioral symptoms when they were diagnosed and started treatment early in the disease rather than later. After three years of follow-up, people who were diagnosed when they had a clinical dementia rating (CDR) of 0.5 were coping better than peers diagnosed at CDR 1. The results imply that early diagnosis might help AD patients live at home longer, Hallikainen said.—Madolyn Bowman Rogers.
If one genome-wide association study yields a handful of new Alzheimer’s genes, then four GWAS combined should provide a double-handful or more. So reasoned the hundreds of researchers who joined forces in the International Genomics of Alzheimer’s Project (IGAP) meta-analysis to pick out genes whose effect size might have been too small to pop up in the individual GWAS. The researchers presented their findings at the Alzheimer’s Association International Conference, held in Boston July 13-18, and expect to publish them shortly. The work turned up 11 new loci that contribute to risk of late-onset AD, reported Jean-Charles Lambert of INSERM in Lille, France. Members of the collaboration are picking apart the GWAS data in various ways. For example, Peter Holmans of Cardiff University in the UK presented AD genomics through the lens of pathway analysis, hunting not for individual genes but for functionally related groups that contribute to risk. He picked out immunity, cholesterol metabolism, endocytosis, and ubiquitination as key modulators of AD.
“These studies are filling in the jigsaw of Alzheimer risk loci, and perhaps more importantly, they point to four pathways that are of interest, ”wrote John Hardy of University College London, UK, in an email to Alzforum (see full comment below). Rita Guerreiro, also at University College London, told Alzforum, “We have new genes that we want to follow, and that is always good, even if they have a very small effect.”
Scientists estimate that people inherit 60-80 percent of their risk for late-onset AD, with the remainder being environmental (see AlzGene). Previous GWAS identified nine loci; together with ApoE, these account for one-quarter of this heritability. That leaves plenty of genes still awaiting identification, and researchers formed IGAP in 2011 to narrow that breach. They pooled GWAS data—encompassing 17,000 AD cases and 38,000 controls—from four different studies. They are conducted by the European Alzheimer’s Disease Initiative, the UK-based Genetic and Environmental Risk in Alzheimer’s Disease collaboration, and two U.S. studies—the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium, and the Alzheimer’s Disease Genetics Consortium. They reasoned that the pooled data would give them greater ability to detect genetic variants with small effects on risk as well as rare variants.
Meta Results
The main IGAP analysis, presented by Lambert, included two stages. From the meta-analysis of the four previous GWAS studies, the researchers identified nearly 12,000 genetic loci potentially related to AD. In the second stage, they tested these with a new analysis of 8,500 cases and 11,000 controls. The result: 20 genes reached statistical significance. Of these, 11 were new hits pointing to loci listed below.
Lambert told Alzforum that the link between genes identified in an unbiased screen and the physiology of dementia is not always obvious at first glance, adding that plenty of research will be necessary to determine how each of these genes contributes to AD.
On the list of 20 genes, one was conspicuously absent—CD33. Though other GWAS have established its link to AD (see Hollingsworth et al., 2011 and Nai et al., 2011), CDC33 only reached statistical significance in the stage 1 IGAP analysis, but disappeared in stage 2. The authors carefully checked for technical issues, such as problems in genotyping or imputation, which corrects for missing data in a person's genotype, but could not explain why CD33 did not make the grade. Lambert said one possible explanation is that CD33 only plays a strong role in certain populations, which must not have been represented in the IGAP stage 2 sample. Both he and Guerreiro agreed that CD33’s absence here does not mean previous studies linking the gene to AD were incorrect. However, Guerreiro said, it does make her wonder what other AD genes have not yet surfaced in GWAS, emphasizing the importance of further studies.
Following Pathways
Many genes may contribute to AD in ways too small to make a noticeable splash in the big pond of a GWAS. Holmans' pathway analysis approach asks whether gene variants that lack statistically significant linkage can still tell researchers something about AD risk. He sorted such genes from the IGAP analysis into different groups based on their involvement in similar biological functions. Then, he looked for gene-function categories that have more IGAP hits than would be expected by chance. Holmans has already applied this kind of analysis to bipolar disorder (Holmans et al., 2009) and Parkinson’s disease (Holmans et al., 2013).
Previously, the group reported a pathway study of AD using two published AD GWAS and at AAIC 2012, Holmans revealed that genes related to endocytosis and cholinergic receptors contribute to risk (see ARF related news story). In Boston, he presented results from the larger IGAP dataset. Four functional modules were enriched in this analysis: ubiquitination, endocytosis, immunity, and cholesterol metabolism. The study marks the first time that ubiquitination has popped up in an unbiased GWAS. None of the genes in that pathway came out in the individual GWAS; it took pathway analysis to identify their importance as a group.
“Peter’s talk was radically innovative,” commented David Brody of Washington University in St. Louis, Missouri. “We did not know that these four major pathways are heavily implicated in the heritability of Alzheimer’s disease.” Much of the value of Holmans’ analysis, he said, rests on GWAS being an entirely unbiased search and on the findings holding up in the new stage 2 sample.
Nonetheless, there are caveats to the pathway approach, Holmans himself said. For one, he was limited by the gene ontogeny categories available in published databases such as the Gene Ontogeny website and the Kyoto Encyclopedia of Genes and Genomes. If no one has created a particular category, then his analysis cannot identify that pathway. For example, APP processing did not come up, despite its link to AD. In addition, Holmans had to be careful that one powerful gene did not dominate a category. Because he was looking for groups of genes all with small influence, Holmans removed heavy hitters like ApoE from the analysis.
Holmans’ results support previous links between these pathways and AD (for example, see ARF related news story on Kurup et al., 2010 and Halawani et al., 2010; ARF related news story; ARF related news story; ARF related news story). They suggest that the pathways could be fruitful targets for therapeutics. Rather than targeting each individual gene in the group, he suggested scientists could look for a major player or biomarker to influence a whole pathway via new treatments.
Looking Forward
IGAP scientists still have plenty to do, analyzing the dataset in different ways and determining how the identified genes relate to AD. At this point, Lambert said, it is difficult to calculate how much of AD heritability results from these 11 new genes. For one, the proper statistical tools to quantify their contribution to AD risk do not yet exist.
Lambert said that eventually GWAS will yield all the genes this approach can identify, which is mainly common variants with small effects. Already, scientists are turning to exome and whole-genome sequencing to identify rarer variants that may be strong risk factors, such as the recently identified Trem2 gene (see ARF related news story; ARF related news story; and ARF related news story).
The IGAP and other GWAS have shown scientists that polygenic diseases incorporate even more genetic risk factors than initially believed, commented Sudha Seshadri of Boston University, who co-chaired the IGAP session at the meeting. While this is perhaps bad news for the future prospect of personalized genomics, where doctors hope to identify disease risk based on genomes, it is good news for scientists aiming to develop treatments, she said. “We are expanding the number of biological targets to study,” Seshadri said.
New IGAP Genes
A region containing HLA-DRB5 and DRB1. These genes encode parts of the major histocompatibility complex II, which presents antigens on the surface of lymphocytes and macrophages. They influence susceptibility to multiple sclerosis (Sawcer et al., 2011) as well as Parkinson’s disease (Nalls et al., 2011).
SORL1 . This sortilin-related receptor is thought to function in endocytosis and cargo sorting. It has long been a suspect in AD (see ARF related news story; ARF related news story on Pottier et al., 2012; Miyashita et al., 2013), but had been controversial because not all studies were able to replicate the association. Now that it has showed up in IGAP, “SORL1 is clearly a good candidate,” Lambert concluded.
SLC24A4, solute carrier family 24, member 4. This gene encodes an ion exchanger involved in pigmentation of skin, hair and iris (Sulem et al., 2007, Duffy et al., 2010). It also appears to affect blood pressure and hypertension (Adeyemo et al., 2009). This role might be the link to AD, where cardiovascular function is a contributing factor, Lambert told Alzforum.
PTK2B, protein tyrosine kinase 2 beta, encodes a cytoplasmic protein involved in ion signaling and long-term potentiation (Huang et al., 2001).
ZCWPW1 contains a zinc finger domain frequently found in proteins that perform epistatic regulation (He et al., 2010).
CELF1 participates in RNA splicing, and possibly editing and translation as well. It may be involved in myotonic dystrophy (Watanabe et al., 2004).
FERMT2, a member of the fermitin family, participates in angiogenesis (Pluskota et al., 2011).
CASS4 codes for a scaffolding protein of unknown function.
INPP5D, inositol polyphosphate-5-phosphatase, dampens blood cell proliferation and survival.
MEF2C, myocyte enhancer factor 2C, encodes a transcription factor involved in muscle development. Mutations in the gene cause intellectual disability (Nowakowska et al., 2010, Le Meur et al., 2010). MEF2C has also been linked to synapstic plasticity (Akhtar et al., 2012).
NME8 encodes a gene implicated in primary ciliary dyskinesia (Zariwala et al., 2007).—Amber Dance.
A patient comes in to the doctor’s office because she can no longer recall cousins’ names, or once got lost in the local mall. Should she, and her doctor, be concerned? Physicians have often dismissed the memory-related complaints of otherwise healthy thinkers as the hypochondria of the “worried well.” But feeling that one’s thought processes are waning could be a sign of something real, according to research presented at the Alzheimer’s Association International Conference (AAIC), held July 13-18 in Boston. “The purely subjective feeling of cognitive decline may be the first sign of future Alzheimer’s disease,” said Frank Jessen of the German Center for Neurodegenerative Diseases in Bonn.
The idea has deep roots but is now growing with a new collaboration to develop standard definitions and approaches to measure subjective cognitive decline (SCD). Researchers at AAIC presented preliminary findings on SCD and suggested that testing for it might identify people at early risk for dementia, who would be ideal candidates for prevention trials. The phenomenon was profiled recently in the New York Times.
Ronald Petersen of the Mayo Clinic in Rochester, Minnesota, who moderated an AAIC press conference on SCD, commented that the new focus on subjective decline fits a trend in which researchers are looking at earlier and earlier stages of disease. A decade ago many scientists refocused their interests from dementia to mild cognitive impairment, and now are extending it to even earlier signs. This push has been spurred in part by disappointing clinical trial results and the suggestion that the earliest-stage patients might benefit from treatment, suggested Andrew Saykin of Indiana University in Indianapolis in an interview with Alzforum.
“This is not a new concept,” commented Rebecca Amariglio of Brigham and Women’s Hospital, but “it has gotten increasing attention.” She noted SCD was included in 1980s descriptions of cognitive decline and dementia (Reisberg et al., 1982, Reisberg, 1986), and in diagnostic criteria for MCI (Petersen et al., 1999). In 2005, researchers published the account of a chess player who reported his own memory loss two years before he fulfilled MCI criteria. The man noticed the decline because he could only plan two moves ahead in the game instead of his usual four (Archer et al., 2005). And an imaging study in 2006 suggested people with SCD have brain atrophy comparable to those with MCI (see Alzforum News story on Saykin et al., 2006).
Defining the Problem
Now that many scientists agree SCD is worthy of study, how should they go about it? SCD is difficult to capture objectively in similar ways that MCI was a decade ago, Petersen noted. The researchers who presented at AAIC all used somewhat different criteria and even names for the condition, such as “subjective memory impairment,” “self-reported cognitive concerns,” “subjective memory complaints” and “subjective cognitive concerns.” “We cannot compare data across studies because everyone is talking about their own concepts,” said Jessen. “There is no consensus of what we mean when we use these terms.” Jessen presented the work of the Subjective Cognitive Decline Initiative, an international working group of scientists who convened in November of 2012 to better describe this condition and propose standard research practices. Jessen expects the group to publish their consensus soon, and offered a preview at the meeting.
Defining SCD might seem like a simple task, Jessen said, but was far from it. Even deciding on the term “subjective cognitive decline” was tricky. The group concluded that the condition must be based on an individual’s own feelings, not on objective cognitive tests. They chose the broad category of cognitive problems to include functions other than memory. And they picked the word decline to indicate a worsening of thought processes. Notably, the label of SCD applies only to people with normal scores on cognitive tests. It does not include anyone with MCI or dementia, nor people whose cognitive problems can be explained by another neurological condition, medications, or drug use. AD biomarkers may be present, or not. SCD is not a requirement for a diagnosis of preclinical AD, nor is it expected to be present in all cases of preclinical AD.
The working group concluded that currently available data are too limited to define specific features of SCD, but they offered guidelines for future attempts to clarify the issue. Specifically, SCD studies should take note of several features of the decline, such as where someone first reports their concerns. For example, people who are specifically asked about memory loss in a research study questionnaire might be different from those who bring up SCD on their own. Another important feature could be undue worry, because some researchers suspect that may mean a greater likelihood of later MCI or dementia. Finally, the initiative recommends that researchers should record whether the decline is in memory or other domains, the age of onset, whether friends or relatives have noticed problems in the individual, and ApoE4 genotype if possible.
While no one can be certain how often SCD will turn into AD, the group suggested several features that might increase the odds. Problems in memory, onset in people over 60, worries about the issue, and feelings that one’s age-matched peers struggle less could be signs of impending AD. Confirmation of decline by an objective informant was another criterion that could indicate increased dementia risk, but this was controversial among the working group, Jessen said. Further indications of preclinical AD could come from the ApoE4 genotype or AD biomarkers such as cerebrospinal fluid tau or amyloid, hippocampal atrophy, amyloid or glucose utilization scans. The list continues to evolve, Jessen said.
Early Data
Presenters stressed that not everyone who thinks they are getting forgetful has started down the road to AD. “As people age, everybody gets a little forgetful, a little slower,” commented Petersen, who is also part of the SCD initiative. While the precise risk that SCD confers remains uncertain, Richard Kryscio, of the University of Kentucky in Lexington, has started to quantify it. In a longitudinal study with yearly follow-up, his group asked 531 people, average age 73, “Have you noticed a change in your memory since you last visited us?” Of these, 296 said yes, an average of eight years after they had entered the study. Of those, 36 went on to develop mild MCI, and 78 progressed to dementia. It took six or more years for the initial, subjective complaint to turn into a medically diagnosed condition. Another 127 of the 296 SCD participants died without developing noticeable cognitive impairment. Kryscio concluded that someone who believes their memory is failing is 2.8 times more likely to become impaired or demented in the future than a person who doesn’t. However, he noted that SCD “is not a guarantee that you will end up with dementia.”
Participants in Kryscio’s research donate their brains to the study. He has seen that people who report SCD and progress to MCI or dementia have severe AD-related plaques and tangles on autopsy, whereas people who reported SCD but never reached an impaired state had moderate AD pathology. This ties in with current thinking in the field that cognitively normal people who have brain amyloid as seen by PET imaging are in the asymptomatic stage of the disease (see Alzforum News story on Jack et al., 2013).
Kryscio’s findings paralleled those SCD initiative member Amariglio. Her group asked 131 cognitively healthy people, aged 65 or older, about their memory function and correlated responses with brain scans carried out with the amyloid marker Pittsburgh compound B. Amariglio reported that the worse someone reported their memory to be, the more amyloid had built up in their brain.
Carriers of the AD risk gene ApoE4, in particular, have reason to be concerned about memory decline. Cécilia Samieri of the Research Center Inserm in Bordeaux, France, examined subjective cognitive concerns as a potential marker for progression in ApoE4 carriers in a nearly 4,000-person subset of the long-running Nurses’ Health Study. The participants underwent cognitive testing four times over a six-year period from 1995 to 2001. They received ApoE4 screening but were unaware of their status at the time.
ApoE4 carriers made up nearly one quarter of the sample. They were more likely than non-carriers to report seven different concerns: recent memory changes, trouble remembering a short list, difficulty recalling things from one second to the next, trouble recalling recent events, struggling to understand verbal instructions, difficulty following conversations or television plots, and difficulty getting around familiar neighborhoods. These problem areas included in the Nurses’ Health Study predate the move to form an official SCD definition. The researchers found that memory decline occurred in ApoE4 carriers who made even one of the seven types of subjective complaint at the study outset. In contrast, decline occurred in non-carriers who made three or more kinds of complaint. Samieri concluded that subjective concerns were a stronger predictor of decline in ApoE4 carriers than non-carriers.
What kind of decline might await people with SCD? Alexander Koppara of the University of Bonn examined changes in episodic and working memory in 2,319 people, aged about 80 years, who were cognitively normal at the study’s outset. Over the following eight years, people with subjective memory impairment saw their episodic memory decline faster than those who perceived no cognitive problems. Those who were specifically concerned about their memory saw the steepest decline. However, working memory was not impaired, at least over the eight years of this study. Koppara suggested that subjective memory impairment and concerns could be an early indicator for AD, and useful for identifying subjects for prevention trials. This fits with literature reports that episodic memory deficits can occur early on, while working memory problems occur later in AD (Weintraub et al., 2012).
Applying the Concept
Petersen noted that, based on the prevalence of brain amyloid in people in their 70s, the upcoming Anti-Amyloid Treatment in Asymptomatic AD (A4) will have to screen three times the number of subjects the trial calls for in order to find people who meet the trial’s inclusion criteria (see Alzforum news story). An SCD questionnaire to stratify people for their likelihood of having brain amyloid buildup, could reduce costs in similar studies, he suggested. Reisa Sperling of Brigham and Women’s Hospital, Boston, told Alzforum that A4 researchers will assess the presence of SCD in enrollees and whether SCD increases their chance of having a positive amyloid scan, but will not treat those participants as a separate cohort. “About 30 percent of older adults are amyloid positive. Maybe we can get that up to 35 percent if we enrich on subjective complaints or family history,” she speculated in an email to Alzforum. “This will be an important learning piece in our screening process.”
The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging is already analyzing whether subjective memory complaint in people who test in the normal range on cognitive tests correlate with biomarkers or have predictive value.
Similarly, the Alzheimer’s Disease Neuroimaging Initiative (ADNI2) will be adding a new cohort of people with “subjective memory complaints,” wrote Michael Weiner of the University of California, San Francisco, in an email to Alzforum. These people were previously left out of ADNI because their complaints made them not “normal,” but they fell short of criteria for MCI. The Initiative expects to enroll 60-80 people in this category, according to Weiner. People with SCD will likely be enthusiastic about joining a study, noted Saykin, who is also part of the SCDI working group. “They are really concerned about what is happening,” he said. ADNI is now reaching out to people who tried to sign up but were eliminated due to cognitive concerns, inviting them to join the new cohort, Petersen told Alzforum. He expects this group will be heterogeneous but might contain a subset who are truly sensing impending cognitive decline.
The rising interest in SCD means physicians should no longer dismiss the concerns of older people who think their memories are faltering. However, there are many other potential causes of memory loss besides impending dementia, commented both Jessen and Creighton Phelps of the National Institute on Aging in Bethesda, Maryland. For example, attention deficit and visual or hearing problems frequently masquerade as memory impairment that interferes with the daily routine, such as following a conversation or a television program, Phelps noted. Moreover, certain other neurological conditions and medications can interfere with normal memory. “You need to eliminate these other things first,” Phelps said.
Amariglio offered examples of normal memory problems during aging that should cause no undue anxiety—such as forgetting why you went into the garage, or struggling with people’s names. More worrisome symptoms might include getting lost in your hometown, struggling to recall the details of recent activities, or noticing a worse memory than others in the same age group. As Phelps put it, if you forget where you left your keys, that’s no big deal, but “if you cannot remember how to use them, it is a problem.”—Amber Dance.
No Available Comments
Part 1 of a two-part story. See part 2.
While Alzheimer’s therapies that target amyloid β production and clearance grab the lion’s share of the headlines, they comprise but a fraction of the strategies under investigation. At the Alzheimer's Association International Conference (AAIC), held July 13-18 in Boston, Massachusetts, speakers laid out approaches ranging from anti-amyloid therapies to cholinergic transmission, inflammation, and regeneration. Some researchers detailed Phase 3 or 2 data, others discussed compounds entering Phase 1, and some approaches have yet to make the jump to human trials (see Part 2 of this series). In line with recent trends in the field, most researchers aim to intervene early in the disease, and some are launching large prevention trials with the goal of delaying symptom onset in preclinical Alzheimer’s disease (AD) populations. Read on for a sample of the clinical trial data presented at AAIC.
Attacking Amyloid
Anti-amyloid strategies have garnered intense interest, but the field is still waiting for a home run in this area. Baxter International Inc. recently announced that its intravenous immunoglobulin (IVIG), Gammagard, missed its primary endpoints in a Phase 3 trial (see ARF related news story), but at an AAIC press briefing, principal investigator Norman Relkin at Weill Cornell Medical College, New York City, was not ready to give up. Relkin fleshed out the prior topline results with preliminary biomarker data. IVIG treatment lowered levels of Aβ42 in the blood and boosted anti-amyloid antibodies in cerebrospinal fluid (CSF), Relkin said. It also thinned out fibrillar Aβ deposits in the brain, as shown by Amyvid PET scans. Relkin said investigators are still analyzing MRI and FDG PET scans to learn about IVIG’s effect, if any, on brain atrophy and metabolism. Those data will come in the fall. “I am optimistic that there are signals worthy of further study,” Relkin told Alzforum, although he added it was too soon to plan another trial.
Planned subgroup analyses showed that people with moderate AD or who carry an ApoE4 allele responded better to the treatment, improving on some cognitive measures. Looking back, the 24 patients in the Phase 2 trial of Gammagard, which had been declared a success, happened to fall into that group, Relkin said. In those phase 2 patients, Gammagard appeared to slow cognitive decline and brain shrinkage over an 18-month extension trial (see ARF related news story). Some of those patients have now been treated for more than eight years. “I cannot tell you they have not progressed, but it is much slower than expected. At least two are still in the mild state of the disease,” Relkin said. AD is normally fatal within eight to 10 years, but some cases of slow progression occur naturally, too.
Other IVIG therapies have recently met with mixed results. Octapharma AG’s IVIG missed its primary endpoints in a Phase 2 trial of AD patients (see ARF related news story), but looks promising so far in a small Phase 2 trial in people with mild cognitive impairment (MCI) (see ARF related news story). Grifols Biologicals, Inc., Sutter Health, and Behring AG in Bern, Switzerland, are also testing pooled antibodies, although they have not reported data yet (see ARF related news story).
Enhancing Cholinergic Transmission
Consider the α7 nicotinic acetylcholine receptor, now gaining luster as an AD target (see ARF related news story; ARF related news story). At last year’s AAIC, researchers reported that EnVivo Pharmaceuticals’ α7 agonist met its endpoints in a Phase 2b trial (see ARF related news story). This year a new player joined the club, to great audience interest. Laura Gault at pharmaceutical company AbbVie, Chicago, Illinois, presented data from a 12-week Phase 2 trial of her company’s α7 agonist ABT-126. AbbVie spun off from Abbott Laboratories in 2012. The trial enrolled nearly 300 participants with mild to moderate AD and an average age of 74. They received either placebo, one of two doses of ABT-126, or the approved AD drug donepezil, an acetylcholinesterase inhibitor. The trial had few dropouts, and adverse events were mild and similar to those seen with donepezil. People complained mostly about headaches and gastrointestinal problems, Gault said. This trial began in 2009, and ended in 2010.
The low dose of ABT-126 had no effect. On the higher dose, participants reached similar cognitive gains to those seen on donepezil, modestly improving their scores on memory tests in the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog) compared to the placebo group. Analysis showed that participants with higher levels of ABT-126 in their blood performed significantly better on the ADAS-Cog than those with less drug exposure. The relationship showed no evidence of a plateau, implying that people could improve more at higher doses, Gault said. Additional toxicology studies in animals and prior Phase 1 studies suggested that higher doses of ABT-126 would be well-tolerated, she claimed. Based on this, AbbVie is running two 24-week Phase 2b studies enrolling about 400 AD patients apiece that will explore higher doses. Gault declined to specify what any of the doses are. The first trial will again compare ABT-126 to donepezil and placebo, while the second will look at whether a combination of ABT-126 and an acetylcholinesterase inhibitor will provide additional benefit. Those trials are set to read out next year.
Dampening Neuroinflammation
Recent research points to a prominent role for neuroinflammation in AD pathogenesis (see ARF webinar; ARF related news story). At last year’s 5th Clinical Trials in Alzheimer’s Disease conference, held 29-31 October in Monaco, Bruno Imbimbo of Chiesi Pharmaceuticals in Rockville, Maryland, reported that the company’s compound CHF5074 dialed down inflammatory signaling from microglia and improved cognition in a 12-week Phase 2 trial run in the U.S. and Italy (see ARF related news story). CHF5074, a derivative of nonsteroidal antiinflammatory drugs, was originally thought to modulate γ-secretase. Animal and cell culture studies have since shown that it acts on microglia to suppress cytokine signaling and stimulate phagocytosis of amyloid deposits, according to data presented at AAIC 2013.
Joel Ross at the Memory Enhancement Center in Eatontown, New Jersey, led the U.S. portion of the trial. The trial enrolled about 100 people with mild cognitive impairment (MCI). Over the initial three months, those who carried an ApoE4 allele improved on several cognitive measures, while non-carriers did not, Ross said at an AAIC press briefing. Most participants continued on drug in an open-label extension phase. Ross said that ApoE4 carriers in the open-label extension have maintained their cognitive gains over 88 weeks of treatment. They score better on several measures of verbal memory, as well as tests of attention and executive function, compared to their baseline performance. Non-carriers showed no immediate benefit, Ross said; however, their cognitive abilities have remained stable for the nearly two years of the extension study. With regard to biomarkers, patients on drug showed linear decreases of inflammatory markers such as TNF-α and sCD40L in CSF.
What might account for the greater effect of CHF5074 in ApoE4 carriers? These people are believed to experience more neuroinflammation than non-carriers, Ross said (see, e.g., ARF related news story ).
Biotech startup CereSpir Inc. announced July 26 that it will license CHF5074 from Chiesi, with plans to develop and commercialize the product (see press release). CEO Daniel Chain founded the company for this purpose. Chain told Alzforum that he hopes to start a Phase 3 trial in ApoE4 carriers in 2014. Participants will either have amnestic MCI or be asymptomatic but with affected parents. Roughly half of AD patients have at least one copy of ApoE4.
Boosting Brain Insulin
Diabetes is a risk factor for Alzheimer’s (see AlzRisk analysis). In small trials, insulin administered through the nose has appeared to help people with MCI or early AD maintain cognition and daily function (see ARF related news story; ARF related news story). However, because of rapid metabolism in the CNS, this treatment results in a short spike of brain insulin. At AAIC, principal investigator Suzanne Craft, now at Wake Forest School of Medicine, Winston-Salem, North Carolina, discussed a Phase 2 trial of a long-acting insulin analogue called detemir. Marketed by the company Novo Nordisk, this compound lasts from 12 to 22 hours in the brain, resulting in a smaller jump in insulin and longer overall exposure. Detemir better mimics basal insulin levels in the brain, Craft noted (see Hennige et al., 2006; Tschritter et al., 2007). The trial was sponsored by the University of Washington and the National Institute on Aging.
Aptly named SNIFF-LONG 21 (Study of Nasal Insulin to Fight Forgetfulness—Long-acting Detemir—21 Days), the study enrolled 60 adults with early AD or amnestic MCI to receive placebo, 20 international units (IU), or 40 IU of detemir. 40 IU is less than 2 milligrams. After three weeks, treatment groups showed no improvement on a verbal memory composite test that served as primary outcome measure. Participants on the higher dose performed better in tests of visual retention and working memory, and a pre-planned subgroup analysis suggested that ApoE4 carriers taking 40 IU of detemir made strides in verbal memory, while non-carriers declined. The enhancement was robust and appeared in the majority of carriers, Craft claimed, although the numbers of ApoE4 carriers in this one subgroup were small. In addition, people whose insulin resistance at baseline was high, as assessed by the Homeostasis Model of Assessment–Insulin Resistance (HOMA-IR) test, scored better on memory tests after three weeks, while those with low baseline insulin resistance declined. The interactions with ApoE genotype and insulin resistance seem to be independent effects, Craft said.
Curiously, the opposite pattern had occurred in prior trials of regular insulin: ApoE4 non-carriers did better on verbal memory tests, while carriers did not (see Reger et al., 2006). The reason for this discrepancy is unclear. To compare the effects of insulin and detemir side by side, Craft is conducting the 4-month Phase 2 trial SNIFF-LONG 120 of 90 people with early AD or MCI. The primary outcome measure will be changes on the ADAS-Cog. Participants receive 40 IU of either insulin or detemir, or a saline placebo. The researchers will measure insulin and detemir levels in CSF to estimate drug exposure, which may provide clues for why only certain groups respond to the treatment, Craft said.
For Phase 1, preventative, and preclinical studies, see Part 2 of this series.—Madolyn Bowman Rogers.
No Available Comments
No Available Further Reading
This concludes a two-part series. See part 1.
No major clinical trial results were announced at this year’s Alzheimer's Association International Conference (AAIC), held July 13-18 in Boston, Massachusetts. Instead, researchers provided updates on investigational therapies. Some presented phase 3 or 2 data (see part 1 of this series), while others outlined approaches that are just entering the clinical pipeline. Here is a sampling of the latter.
Stimulating Regeneration
Could regenerative approaches preserve cognition in a disease that, after all, is marked by rapid loss of neurons and brain mass? Speaking at an AAIC press briefing, Roberta Brinton from the University of Southern California, Los Angeles, said that the neurosteroid allopregnanolone may do just that. Allopregnanolone is a metabolite of the hormone progesterone. It is made in the brain and spinal cord, and occurs at high levels in the blood of pregnant women. In the triple transgenic mouse model of Alzheimer’s disease (AD), the steroid promotes the birth of new neurons and rescues cognition, while lowering amyloid β pathology and dampening inflammation, Brinton said.
Brinton and Lon Schneider, also at USC, will lead a Phase 1 trial of allopregnanolone in 16 men and 16 women with mild cognitive impairment (MCI) or early AD. Participants will receive intravenous infusions of the drug once per week for three months. To assess safety and tolerability, the trial will test multiple ascending doses of the steroid, with the highest dose sufficient to boost blood levels to those seen during pregnancy. Brinton expects the trial will be funded by the National Institute on Aging and conducted through the USC Alzheimer’s Disease Research Center, and will begin next year. Michael Rogawski and Gerhard Bauer at the University of California, Davis, will supply the drug and placebo control.
As secondary endpoints, the researchers will compare baseline and final MRI scans of hippocampal volume, white matter hyperintensities, and brain connectivity. Participants will also take standard neuropsychological and cognitive tests. If the drug proves safe in Phase 1, Phase 2 studies will look for a slowing of cognitive decline and brain atrophy over 12 or 18 months, Brinton said. In addition, Rogawski is currently testing allopregnanolone in people with traumatic brain injury.
Earlier studies showed that allo, as the drug is known for short, kindles neurogenesis. It opens chloride channels in neural stem cells, leading to depolarization, increased calcium levels, and transcription of neuronal differentiation factors (see ARF related news story; ARF related news story). Treatment once per week appears to be optimal, because more frequent dosing causes a feedback loop that shuts neurogenesis back down, Brinton told Alzforum (see Chen et al., 2011; Brinton et al., 2013). Because the hormone regulates multiple cellular systems, it also affects diverse processes such as inflammation, cholesterol trafficking, and Aβ production. In 3x Tg mice, treatment with this steroid was reported to rescue associative learning and memory deficits, and to increase mitochondrial respiration, resulting in greater energy production, among other effects (see Irwin et al., 2011; Singh et al., 2012).
Prevention With Pioglitazone
With accumulating evidence that the pathogenesis of AD begins twenty years or more before diagnosis, many researchers are interested in identifying healthy populations at high risk for AD and testing interventions that might delay symptoms. At an AAIC press briefing, Kathleen Welsh-Bohmer from Duke University, Durham, North Carolina, described one such approach. The Phase 3 Tommorrow Study (no, that is not a misspelling) will enroll 5,800 cognitively normal people at 50 sites in the U.S., Europe, and Australia. Participants must be at heightened risk for AD based on age and the genetic factors ApoE4 or Tomm40 variant, hence the study’s name (see ARF related news story). Half the participants will receive a low dose of the approved diabetes drug pioglitazone, which will be supplied by the trial’s sponsor, Takeda Pharmaceuticals (see press release). The Tommorrow trial will start in August and run for four years, with the primary outcome measure being conversion to MCI. In that amount of time, the researchers expect to see 410 conversions, which will give them 90 percent power to detect a treatment effect, Welsh-Bohmer said.
Why pioglitazone? This PPAR-γ agonist improves cognition in AD mice, but had mixed results in prior human trials (see Miller et al., 2011). Researchers involved in this and other human trials noted that pioglitazone and related thiazolidinediones seem to improve brain metabolism, and suggested they might help patients in early stages of the disease (see ARF related news story). Meanwhile, animal studies continue to turn up novel mechanisms for how these drugs might restore cognition and lower pathology (see ARF related news story). Collaborator Allen Roses at Duke told Alzforum that the choice of pioglitazone was guided by the drug’s excellent safety track record, as well as human and animal trial data suggesting that the drug dampens inflammation, curbs amyloid pathology, and improves brain blood flow and use of oxygen and glucose (see, e.g., Sato et al., 2011).
Welsh-Bohmer noted challenges in designing a global preventative study. For one thing, the researchers needed to find cognitive tests that are valid worldwide and are sensitive to changes in the MCI population. Welsh-Bohmer, working with a group of international AD clinical trial experts, selected an outcome battery that includes tests of episodic memory, executive function, language, attention, and visuospatial skills. The researchers are currently validating the test battery in 200 cognitively normal people 65 to 88 years old in Italy, Russia, and Switzerland. They will compare this group’s performance against that of 25 people with Alzheimer’s disease. The goal is to determine what constitutes normal performance across different cultures and a range of ages. The researchers also had to define criteria for MCI. They chose a clinical dementia rating (CDR) of 0.5, plus failing two of 12 measures on the test battery on two consecutive exams taken six months apart. Designing the trial took two years of literature reviews and modeling, Welsh-Bohmer said. She suggested that this work could lay the groundwork for future trials in preclinical AD populations.
Prevention Through Improved Health
Another prevention study takes a different tack. Speaking in an AAIC plenary session, Karen Ritchie from the public research institution INSERM in Paris, France, discussed epidemiological factors that increase risk for AD. The Esprit Project in Montpellier, France, enrolled almost 2,000 cognitively normal people over age 65 and followed them for 12 years, tracking cognitive scores and conversion to MCI or AD (see ARF related news story). The goal was to quantify the contribution of various environmental or behavioral risk factors to AD risk. The data suggested that increasing education could lower AD incidence by about a quarter, while controlling conditions such as diabetes, depression, and stroke could cut another quarter of new cases. Exposure to these risk factors occurs mostly in middle age, Ritchie emphasized. “AD is a disease of middle age that leads to a terminal dementia stage in old age,” she said.
Based on this data, the U.K. Alzheimer’s Association is conducting the PREVENT study, which will investigate whether lowering these risk factors can in fact delay the onset of AD (see also Ritchie and Ritchie, 2012). PREVENT will enroll 600 middle-aged children of people with AD, who are assumed to be at increased risk of developing the disease as they age. In a randomized controlled trial, half the participants will receive an intervention consisting of improved diet and physical exercise as well as computer tasks to increase cognitive reserve. The researchers will track markers of insulin resistance, inflammation, and depression in this group, so that the participants can receive preventative medical treatment at the earliest stage of these disorders. The control group will receive annual checkups and standard medical care from their family practitioner.
The researchers will use cognitive tests and CSF biomarkers to detect the development of preclinical AD or MCI. They will also compare the treatment group to an observational cohort of people at low risk for AD, who do not carry the ApoE4 risk factor or have an affected parent. The hope is that the interventions might lower AD incidence in participants to levels seen in the low-risk group. At the earliest sign of benefit, all participants will be switched to the active prevention group, Ritchie told Alzforum. Several other large European studies are also investigating whether lifestyle changes can reduce AD risk (see ARF related news story).
Epigenetics and Gene Expression
The aging brain undergoes many changes. In primates, genes involved in neural plasticity become less active with age. This is due to changes in chromatin structure that pack DNA more tightly and prevent these genes from being transcribed, said Li-Huei Tsai from MIT in an AAIC plenary session. In animal studies, Tsai identified the enzyme histone deacetylase 2 (HDAC2) as a key regulator to silence genes involved in synaptic plasticity and learning (see ARF related news story; ARF related news story). HDAC2 levels are elevated in AD brains, and neurotoxins such as Aβ induce its expression, Tsai found (see ARF related news story).
Blocking HDAC2 might therefore help restore cognitive function in people with AD, Tsai suggested. She and colleagues have identified a small molecule, SC-027, that inhibits class I HDACs including HDAC2 in a long-acting fashion. The patent on this compound has expired, Tsai said. After 10 days of treatment with SC-027, CK-p25 AD mice have better memory and more synapses than untreated controls, Tsai reported. Three-month-old >5xFAD mice treated for one month have less amyloid pathology in their brains than do controls. It is not yet clear how the inhibitor reduces pathology, Tsai said. When treated at six months of age, the 5xFAD mice learn better than untreated animals. SC-027 also normalizes gene expression in the 5xFAD animals, dialing down genes involved in inflammation and cell death. Tsai said she is interested in taking SC-027 to human trials, but many challenges remain, including toxicity at high doses.—Madolyn Bowman Rogers.
No Available Comments
New research suggests that the innate immune system affects cognition by regulating synapse formation and maintenance. What role might this process play in Alzheimer’s, where immune cells become activated? At the Alzheimer's Association International Conference (AAIC), held July 13-18 in Boston, Massachusetts. Qiaoqiao Shi, working with Cynthia Lemere at Brigham and Women’s Hospital, Boston, reported that AD mice lacking a key component of the complement system have worse amyloid β (Aβ) pathology, but better memory, than their intact littermates. It is not yet clear why, but intriguingly, the mice with deficient complement systems retain more neurons and synapses as they age than their immune-competent brethren. In humans, brain amyloid load correlates poorly with cognition, whereas synaptic density shows a tight relationship with mental skills (see ARF related news story; DeKosky and Scheff, 1990).
The complement cascade comprises more than 20 small proteins that trigger microglia to gobble up harmful debris, such as Aβ deposits. In prior studies, researchers have found that AD mice lacking the central complement component C3 develop more Aβ pathology and neurodegeneration than do controls (see ARF related news story; ARF related news story). However, the complement also plays a role in maintaining synapses, with the C3b subunit cueing microglia to prune unneeded ones (see ARF related news story). This suggests that the complement system could contribute to the aberrant synapse elimination seen in AD, or even in normal aging. Intriguingly, complement components are more abundant in AD and Down’s Syndrome brains than in healthy controls, Lemere said. In addition, previous studies showed that the complement represses adult neurogenesis (see ARF related news story), which may contribute to new circuitry and synaptic plasticity.
To first investigate the role of the complement in aging, Shi, in collaboration with Beth Stevens at Boston Children’s Hospital, compared 16-month old C3 knockout and C3 receptor (CR3) knockout mice to age-matched controls. Both types of knockout had more neurons in the CA3 region of the hippocampus, and more synapses in several brain regions including CA3, dentate gyrus, and visual cortex, Shi reported at AAIC. (Other brain regions, such as the CA1, showed no difference between the knockouts and wild-type.) Hippocampal slices from the knockouts displayed stronger long-term potentiation, and both knockout strains performed better in the water T maze and in contextual fear conditioning trials than did aged controls.
Shi then crossed the knockouts with APP/PS1dE9 AD mice. As expected, the C3-deficient APP/PS1 offspring had more Aβ deposition in cortex and hippocampus at 16 months of age than did their APP/PS1 littermates. Shi traced this to poor phagocytosis of Aβ and fewer reactive astrocytes (see Fu et al., 2012). Despite the increased amyloid load, the C3-deficient APP/PS1 mice outperformed their APP/PS1 littermates with intact complement in the water T maze, learning as quickly as wild-type. Like the wild-type C3 knockouts, the complement-deficient APP/PS1 crosses also had more neurons in CA3 than did their APP/PS1 littermates.
The researchers do not yet know why the crossed mice learn better despite bearing more brain amyloid. It is possible that C3 mediates the toxic effects of Aβ, or that the brain can tolerate amyloid as long as it does not lose synapses, Lemere said. In people, education and cognitive activity early in life seem to build up a “cognitive reserve” that helps protect against Alzheimer’s disease for some time (see ARF related news story; ARF related news story). It also remains unclear why the effects of C3 deficiency vary by brain region and with age. More research is needed to tease out the mechanisms behind the sharper cognition, Lemere told Alzforum.
Lemere said the next step is to see if knocking down C3 after the brain has fully developed, or specifically in the hippocampus of aged AD mice, will be as beneficial as knocking it out from birth. If so, C3 inhibition might become a potential AD therapeutic approach. Small molecule inhibitors of human C3 exist. One was evaluated in a clinical trial for macular degeneration, since the complement system damages photoreceptors in that disease (see Qu et al., 2013; Whitcup et al., 2013).—Madolyn Bowman Rogers.
No Available Further Reading
Having surveyed the genome for common variants associated with Alzheimer’s disease, geneticists are now turning the search lights on rare, hard-to-find mutations that may be even stronger risk factors. At the Alzheimer’s Association International Conference (AAIC), held July 13-18 in Boston, researchers presented potential, if unconfirmed, candidates, including phospholipase D3, the ubiquitin ligase TTC3, and genes that play roles in axon guidance and microtubule stabilization. Scientists report some success sequencing the exomes, or protein-coding regions, of genes from families hard hit with AD. They are thus far having less luck with a separate method called exome chip technology. It searches for rare variants by skimming the genome’s surface, genotyping specific, known mutations in large numbers of people. Researchers at AAIC agreed that at this point, identifying and validating rare variants remains challenging.
Nonetheless, this approach is slowly beginning to pay dividends. Last year, two research groups discovered that rare mutations in the immune cell receptor TREM2 confer as much risk for AD as ApoE4 (see Alzforum News story on Guerreiro et al., 2013 and Jonsson et al., 2013). None of the new variants discussed at AAIC have yet reached TREM2’s status as a definite risk factor, commented Adam Naj of the University of Pennsylvania in Philadelphia. “Everybody was presenting preliminary work that needs a lot of replication,” said Naj, who co-chaired one of two sessions on rare variants at the conference.
Researchers are taking two main approaches to find these rare variants. Some identify families where AD affects several members, and then completely sequence their exomes to find variants that co-segregate with the disease. Others genotype large groups of cases and controls with exome chips that identify known single nucleotide polymorphisms (SNPs). The chip approach saves money compared to deep sequencing many samples, but it cannot identify variants beyond the known SNPs imprinted on the chip. To date, the family approach looks more promising, said Carlos Cruchaga of Washington University in St. Louis, Missouri. One reason is that exome chips still have some technical problems, including high rates of false positive and false negative hits.
Whichever method they choose, researchers looking for rare variants have to do a lot of sifting to find the truly interesting ones. Each person totes thousands of SNPs, and the first-pass analysis will likely identify several that appear linked to disease. “The problem is not finding rare SNPs, it is figuring out which ones are actually doing anything,” said Mark Logue of Boston University. “The trick is prioritizing them for follow-up.” Scientists can do this by checking whether a mutation is likely to have functional consequences, for example if it alters protein structure, or if the nucleotide in question is conserved in evolution, which would indicate an important role in biology.
Debuting Candidates
Here is a taste of the preliminary findings discussed in Boston:
Performing exome sequencing on 14 families, Cruchaga’s team turned up a variant in the phospholipase D3 gene. It appeared in people with Alzheimer’s from two of the clans. In further genotyping of 10,000 cases and controls, the researchers confirmed the variant associated with AD. Sequencing of the gene in a further 4,000 individuals turned up other mutations in the gene that may affect AD risk. Cruchaga suspects that phospholipase D3 could turn out to have as strong an effect as TREM2. However, little is known about the protein’s function, he told Alzforum.
Martin Kohli, who works with Peggy Pericak-Vance at the University of Miami, Florida, presented another family-based study. One of six kindreds examined led him to mutations in the gene tetratricopeptide repeat domain 3. TTC3 is a ubiquitin protein ligase that is crucial for neuron survival (see image below). The AD-linked variant was absent from 10,000 control alleles. Pericak-Vance found the candidate intriguing because of its function in survival signaling pathways, which could easily be linked to dementia. Moreover, it is located in the “Down’s syndrome critical region” on chromosome 21, Pericak-Vance said. Some people with Down’s syndrome have a triple dose of not the entire chromosome 21, but just this fragment. Because most people with Down’s syndrome develop Alzheimer’s, researchers had suspected that some AD-linked gene was hiding in that critical region, Pericak-Vance said. She suggested TTC3 might be it. APP resides outside of this region on chromosome 21, but the critical region includes the BACE2 gene.

TTC3 helps degrade proteins.
[Image courtesy Martin Kohli, University of Miami, Florida, and Developmental Cell]
Robert Graham of Genentech, in South San Francisco, California, reported on exome and whole-genome sequencing of another extended family. This project identified a rare variant that Graham said was more likely to occur in cases than controls in a separate, larger series of subjects. This mutation lies in the gene unc-5 homolog C. UNC5c is one of a family of receptors for netrins. Netrins work during neural development to regulate axon guidance. The researchers found that the mutation reduced cell viability in vitro.
Other researchers have adopted different strategies. Logue, who works in Lindsay Farrer’s laboratory at Boston University, chose to study an ethnic group, that is, African Americans. This population has been under-studied and could offer new insight into genetic risk factors, Logue said. He began by sequencing the exomes of eight African Americans who have a family history of AD. From there, he identified 63 prime suspect SNPs, which he genotyped in a further 435 cases and 422 controls. He discovered 11 cases who had one or two variants in a gene called A kinase anchor protein 9 (AKAP9). One control possessed a rare variant in AKAP9 as well. The AKAP9 protein participates in microtubule stabilization, as does the AD-related protein tau.
The Alzheimer’s Disease Genetics Consortium (ADGC) has pursued the exome chip approach. Li-San Wang of the University of Pennsylvania, Philadelphia, reported that after more than 16,000 subjects having been genotyped, no novel variant has yet reached statistical significance for an association with AD. A larger sample will have to be tested, suggested Wang. One interesting candidate was mucin 17 (MUC17), Naj told Alzforum. Mucins decorate the membranes of epithelial cells where they participate in structure maintenance and signaling.
Rare Findings
Going forward, geneticists will continue to be hampered by the very rarity of what they seek. When scientists find a SNP in a gene in a single family, how can they confirm its association with disease? It might require tens of thousands of individuals to find another with that same variant. The SNP might even be a “private mutation,” unique to one individual or kindred.
Scientists predict they can get around this difficulty by looking for cases of multiple variants in the same gene. In addition, if they cannot find more people with the variant of interest, they can turn to the lab and study the gene in cells and models systems, looking for clues that it might contribute to brain function.
Because it takes so many subjects to find and confirm rare SNPs, scientists are working together in large collaborations. For example, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) is performing whole-genome sequencing, which should be completed imminently, said Robert Green, Harvard University, who works on the project. Members of the International Genomics of Alzheimer’s Project (IGAP) plan to pool their exome chip data. Part of the 2012 Presidential Initiative against AD (see ARF related news story) includes funds for the Alzheimer’s Disease Sequencing Project (ADSP). The project’s first phase, already underway, is to sequence the genome of 582 people from 111 families with high rates of AD. Researchers expect the earliest results from this venture in the next year. In its second phase, ADSP will sequence the exomes of 5,000 AD cases and 5,000 controls.
Cruchaga, Logue, and Wangall predicted plenty more rare variant news in the next few years. Alison Goate of Washington University in St. Louis, Missouri, summed up the attitude of AD geneticists. “Time will tell as to whether any of these findings prove to be replicable and real associations,” she told Alzforum.—Amber Dance.
No Available Comments
This is part 1 of a two-part series. See Part 2.
Proposed research criteria for AD employ biomarkers to identify several stages of preclinical disease, and since their publication in 2011 the field has been pushing hard to test how well they work. At the Alzheimer's Association International Conference (AAIC), held July 13-18 in Boston, Massachusetts, researchers presented data that support the criteria’s usefulness and reinforce current models of biomarker progression. In many cases, different study populations revealed remarkable agreement on the prevalence of each proposed stage of preclinical dementia, and how quickly it advances to the next. At the same time, the data also confirm that substantial numbers of people present puzzling biomarker profiles, indicative of neurodegeneration and clinical AD without detectable amyloid deposits (see Part 2 of this series). Speakers agreed that larger studies will be needed to fully elucidate the relationship between biomarkers and prognosis, as most studies to date have been small.
Research criteria commissioned by the National Institute on Aging and the Alzheimer’s Association distinguish three advancing stages of preclinical AD (see ARF related news story on Sperling et al., 2011). Stage 1 applies to people who have only amyloid accumulation; stage 2 to people with amyloid plus a marker of neuronal injury; and stage 3 to people who in addition have signs of subtle cognitive change. These divisions reflect current models of biomarker staging (see ARF webinar; ARF related news story). More recently, Cliff Jack and David Knopman at the Mayo Clinic in Rochester, Minnesota, suggested two additional stages to the model: stage 0, in which all biomarkers are normal, and “suspected non-AD pathophysiology” (SNAP), marked by neurodegeneration in the absence of amyloid. These five stages encompass 97 percent of cognitively healthy older adults, they reported (see image below and Jack et al., 2012; Knopman et al., 2012).

Stages of preclinical AD Image courtesy, Cliff Jack, Mayo Clinic, Rochester, Minnesota
Biomarkers Predict Impairment in Cognitively Normal Seniors
How well does the model work in practice? At AAIC, Stephanie Vos from Maastricht University in the Netherlands described her work testing the model in a long-running study of 311 cognitively normal elderly people seen at the Knight Alzheimer’s Disease Research Center at Washington University in St. Louis, Missouri. Participants were older than 65 and had a clinical dementia rating (CDR) of 0 at baseline, indicating normal cognition. Vos assessed amyloid pathology and neurodegeneration as measured by cerebrospinal fluid (CSF) Aβ and tau, respectively. At the beginning of the study, 42 percent of the population fit stage 0, 15 percent fit stage 1, 12 percent stage 2, and 4 percent stage 3 criteria. Almost a quarter of the population fell into the SNAP group, and about 5 percent fit none of the categories, Vos reported. These percentages are almost identical to those reported by the Rochester Mayo clinic in a separate population of cognitively healthy older adults using imaging measures (see Knopman et al., 2012), as Knopman noted during the discussion.
Importantly, Vos and colleagues next examined whether this initial staging then helped predict who would develop cognitive problems. They found that over an average of four years, people at advanced stages were more likely to progress to symptomatic AD, defined by a CDR of 0.5 or more, that clinicians believed was due to Alzheimer’s. More than half of those at stage 3 progressed, but only a quarter of the stage 2 group, and 13 percent of stage 1. This suggests that the stages have prognostic value, Vos told Alzforum. Cognition remained stable in the stage 0 cohort.
Cognition declined in only six percent of people who were classified as SNAP. The relative stability of this group in this study cohort contrasts sharply with what other researchers report in cognitively impaired populations, where people classified as SNAP often progress rapidly (see part 2 of this series). Vos suggested that cognitively normal people with stable SNAP may represent a different group than cognitively impaired patients who are negative for amyloid and positive for neurodegeneration. She noted that in the St. Louis cohort, the few SNAP individuals who progressed to AD did have some amyloid pathology at baseline, but it fell below predetermined cutoffs.
Other researchers presented similar data supporting the idea that brain amyloid heralds future AD in people with normal cognition. William Klunk from the University of Pittsburgh, Pennsylvania, noted that although the presence of amyloid β deposits does not always correlate with low cognition at baseline, so far all studies have found a connection between brain amyloid and future cognitive decline. In a study that followed 63 cognitively normal subjects, average age of 75, for about four years, one third of those with brain amyloid developed mild cognitive impairment (MCI) due to Alzheimer's disease, a rate of seven percent per year, he reported. By comparison, of those without detectable brain amyloid, less than three percent per year became impaired. The data support the hypothesis that amyloid burden carries a strong risk of future cognitive decline, Klunk said. In contrast, age, cognitive test scores, and brain hypometabolism did not predict progression in this study.
The progression rate Klunk and Vos found for Aβ-positive participants matches that reported by other groups. For example, Christopher Rowe from Austin Hospital, Melbourne, Australia, said that in the Australian Imaging, Biomarkers, and Lifestyle (AIBL) Flagship Study of Ageing, cognitively normal seniors with brain amyloid developed MCI at a rate of about eight percent per year. This is consistent with data from longitudinal studies showing that people deposit amyloid for 10-15 years before becoming symptomatic, Rowe noted (see ARF related news story).
Klunk also looked at how quickly cognitively normal older adults go from amyloid negative to amyloid positive, i.e. from stage 0 to 1. He reported a rate of 10 percent per year in the 75-year-old cohort. During the discussion, Jack noted that he sees a similar figure of about 13 percent per year in the Mayo Clinic population. The AIBL study previously found that it takes about 12 years to go from a negative amyloid scan to the threshold for a positive scan, which agrees well with these figures (see ARF related news story).
Researchers at the meeting asked whether this means that all older people will eventually become amyloid positive. Klunk speculated that they might if everyone lived to be over 120, yet there seems to be large variation in when amyloid deposition starts in different individuals. Klunk added that this does not necessarily mean that everyone would develop clinical AD, since other brain changes must occur before cognition starts to fail.
Memory Impairments and Brain Atrophy Foreshadow Dementia
What about people who already have cognitive problems? People diagnosed with MCI represent a mixed group, researchers agreed. Not everyone will go on to develop Alzheimer’s dementia. Some have underlying pathologies that are not AD, and their cognition may remain stable or even revert to normal. Which biomarkers are most prognostic? Here too, amyloid burden is central but not the whole story, as brain atrophy and other markers of neurodegeneration may be even more predictive, according to research presented at AAIC.
Rowe reported that in the AIBL study, 86 percent of people with amnestic MCI and brain amyloid progress to Alzheimer’s dementia over a period of three years. The data support the concept that MCI with a positive amyloid scan represents prodromal AD, Rowe told Alzforum. Other speakers described similar findings indicating that memory loss plus amyloid strongly predict an Alzheimer’s dementia diagnosis within three to five years, contributing to the emerging consensus in the field.
Both clinicians and patients tend to be unsatisfied with the “three to five year” time window often invoked in research studies. Is there a way to time progression more precisely? In current staging models, amyloid accumulates for two to three decades before diagnosis, whereas markers of neurodegeneration such as rising tau and a shrinking brain appear only a few years before symptoms. David Wolk at the University of Pennsylvania, Philadelphia, hypothesized that these neurodegenerative markers might predict short-term progression better than does the presence of amyloid. Wolk and Brad Dickerson at Massachusetts General Hospital previously defined an “AD signature.” This is a pattern of cortical thinning in nine AD-vulnerable brain regions that discriminates people with mild AD from controls (see ARF related news story; Dickerson et al., 2009). Since brain volume also shrinks with age (see Bakkour et al., 2013), Wolk and Dickerson have now adjusted this signature to remove the effects of aging, Wolk said at AAIC.
At AAIC, Wolk presented data from a cohort of 156 MCI patients enrolled in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). As expected, people who progressed to AD within one year had significantly worse baseline-adjusted AD signatures compared to people who progressed in three years. Both groups had more cortical thinning than people who remained stable over three years. Supporting Wolk’s hypothesis, one- and three-year converters displayed similar levels of Aβ in the CSF.
Recent studies from the Netherlands also found that high CSF tau and brain atrophy, but not amyloid, predict rapid development of dementia (see van Rossum et al., 2012; van Rossum et al., 2012).
The results suggest that brain atrophy can be used to predict how quickly someone might develop AD, whereas amyloid cannot, Wolk concluded. “Neurodegeneration better tracks cognition and your likelihood of having symptoms in the future,” he told Alzforum. —Madolyn Bowman Rogers.
No Available Comments
This is part 2 of a two-part series. See Part 1
Since the advent of brain amyloid imaging, researchers have been surprised to find that a substantial minority of people clinically diagnosed with Alzheimer’s disease have no detectable amyloid β deposits in the brain. In some large trials, up to one third of the ApoE4-negative ‘AD’ patients also turned out to be negative on amyloid scans (see, e.g., ARF related news story). “Amyloid-negative neurodegeneration is more common than we would have suspected,” David Wolk, University of Pennsylvania, Philadelphia, told Alzforum at the Alzheimer's Association International Conference, held July 13-18 in Boston, Massachusetts.
What do these people have? Researchers at AAIC presented the latest on these patients. They seem to represent a heterogeneous group. In some studies, half of them show an AD-like pattern of neurodegeneration on MRI scans and analysis of fluid biomarkers, and progress rapidly from cognitive impairment to dementia. In other studies, amyloid-negative participants seem to have a milder, slower-progressing disorder than AD, with distinct patterns of neurodegeneration. Neurodegeneration in the absence of amyloid has been dubbed “suspected non-AD pathology” (SNAP). While the underlying pathologies remains mysterious, researchers agree this will be an important group to understand.
In Boston, Wolk described how about one-quarter of patients with mild cognitive impairment (MCI) in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort (see Part 1) had no sign of brain amyloid. Of these, 40 percent tested positive for markers of neurodegeneration such as elevated cerebrospinal fluid (CSF) phospho-tau and an AD-like pattern of cortical thinning. Of these 16 patients, four, or 25 percent, developed clinical AD in one year, compared to a rate of 4 percent among people without amyloid or markers of neurodegeneration. This is similar to rates reported by the Mayo Clinic for their SNAP group, (see Petersen et al., 2013). “It suggests that amyloid doesn’t tell the whole picture with regard to development of dementia,” Wolk told Alzforum.
Other studies show similar rates of progression. Consider the Australian Imaging, Biomarkers, and Lifestyle (AIBL) Flagship Study of Ageing. Of its 87 participants with MCI who had MRI and PiB PET brain scans, 28 were amyloid-negative. Eight of them developed dementia in three years, and five of these eight appeared to have AD. None were ApoE4 carriers, reported Christopher Rowe from Austin Hospital, Melbourne, Australia. Rowe did not divide the amyloid-negative group into people with and without neurodegeneration, but he did find that having hippocampal atrophy increased the odds of developing AD. By contrast, 77 percent of amyloid-positive participants progressed to AD within three years.
What might be happening in amyloid-negative patients? One possibility is that they have a non-AD dementia that resembles Alzheimer’s clinically, such as frontotemporal dementia or Dementia with Lewy Bodies. However, Wolk pointed out that in ADNI, the pattern of brain shrinkage and its relationship to CSF phospho-tau resembled that of AD, making it less likely that they have a non-AD dementia. He suggested they might have amyloid that current PET tracers cannot detect. PET ligands bind to fibrillar amyloid deposits but not, for example, to diffuse amyloid or soluble Aβ oligomers, which are widely believed to be more toxic. There is also discussion of amyloid strains.
At AAIC, Brad Dickerson at Massachusetts General Hospital, Boston, hinted that tau pathology underlies some cases of amyloid-negative MCI. He reported that in a cohort of 167 MCI patients, higher CSF tau correlated with cortical thinning, particularly in the medial temporal lobe where shrinkage associates with memory loss. In MCI patients without brain amyloid, neurofibrillary tangles in the medial temporal lobe accounted for memory impairments, Dickerson said.
Other Theories on SNAP
Hyo Jung Choi at Seoul National University Hospital, South Korea, proposed that apolipoprotein A1 could play a crucial role in SNAP. ApoA1 is the main component of high-density lipoprotein (HDL), aka the “good cholesterol.” Choi followed 28 people with amnestic MCI for one year. About half of them had a positive amyloid brain scan, and in this group, about 25 percent developed dementia. Nobody who was amyloid-negative progressed within the year. This subset had low serum ApoA1, which correlated with poor cognition and a distinct pattern of brain atrophy. The pattern showed more shrinkage in temporal and frontal lobes and less in medial temporal lobe, compared to brain atrophy in people who did have amyloid. This suggests that a distinct degenerative process occurs in people without brain amyloid, Choi said.
How might ApoA1 contribute to degeneration? ApoA1 has antioxidant properties and can attenuate the inflammation caused by microglia and astrocytes. Therefore loss of ApoA1 might exacerbate neuroinflammation, Choi suggested. In AD mouse models, too little ApoA1 leads to greater buildup of amyloid in blood vessels and poorer cognition, while abundant ApoA1 protects against these problems (see ARF related news story).
Vascular damage in the brain can damage cognition independently of amyloid burden (see ARF related news story). Duygu Tosun from the University of California, San Francisco, examined diffusion-weighted MRI scans, which show axonal integrity, from a small group of MCI patients in ADNI2 and ADNIGo. Amyloid-negative patients had damage in their fornix, the bundle of fibers that connect the hippocampus to the hypothalamus, compared to amyloid-positive patients and controls, she reported. These patients also had more white matter lesions and vascular damage than controls. Moreover, they showed a different pattern of brain atrophy and hypometabolism than the amyloid-positive group. All told, her data suggest that amyloid-negative MCI might represent vascular cognitive impairment, Tosun said.
At this point, the amyloid-negative group appears to represent a mixture of several different types of pathology, researchers agreed. Most of the studies to date have been small, and larger samples will be needed to parse the reasons for amyloid-negative cognitive impairment, they concluded.—Madolyn Bowman Rogers.
In line with recent analyses in the U.S. and Europe, a July 16 paper in the journal Lancet reported a substantial drop in dementia prevalence among older adults in England over the last two decades. According to the new research, dementia rates fell 24 percent—from 8.3 to 6.5 percent of seniors aged 65 years and older—between 1991 and 2011. “This study was designed to test for differences across sites and across time by using methods that allow comparisons with the power to detect such differences. It is the only study that does this,” noted senior investigator Carol Brayne, Cambridge Institute of Public Health, UK, in an email to Alzforum. The data received wide attention at the Alzheimer’s Association International Conference held July 13-18 in Boston, Massachusetts, and in lay media outlets (e.g. see New York Times and National Public Radio). Alzforum recently covered this trend in depth (see ARF related news story).
Estimates of 1991 dementia rates in England came from the country’s first Cognitive Function and Ageing Study (CFAS I)—a two-stage process with screening followed by diagnostic assessment. The first phase of CFAS I analyzed data from 7,635 seniors at residential home and care facilities in Cambridgeshire, Newcastle, and Nottingham. They were interviewed between 1990 and 1993 about their lifestyle, health, cognition, and sociodemographic characteristics. The investigators selected 20 percent of those people, representing a broad spectrum of cognition, to take the geriatric mental state (GMS) examination. From this they estimated dementia prevalence using a computerized algorithm. CFAS II similarly analyzed data collected between 2008 and 2011 from a near identically sized cohort from the same geographic areas; the only difference was that screening and assessment occurred in one phase.
Age- and sex-specific prevalence estimates from CFAS I put the number of demented elderly in England at 664,000. Population aging effects would increase that figure to 884,000, the researchers report. However, CFAS II found that only 670,000 seniors had dementia in 2011—214,000 (24 percent) fewer than predicted by population aging.
To account for possible skewing due to difficulty enrolling participants, the researchers tested various scenarios that could have nudged the 2011 prevalence figures upward. For instance, they re-calculated the figures assuming that non-responders’ dementia risk was 50 percent, or even double, that of study participants. These scenarios did increase prevalence estimates, but not to 1991 levels, lending credence to the drop in dementia rates in the past two decades.
Other research has also hinted at declining dementia burden in high-income countries. Recent research from Sweden suggests that dementia incidence, or the number of new cases per year, has plunged as public health improved (Qiu et al., 2013). In this study, dementia prevalence hardly budged, though the study was smaller and had a shorter time period than the UK analysis, Brayne noted. Dementia incidence seems to be on the wane in the Netherlands (Schrijvers et al., 2012) and the U.S. (see Rocca et al., 2011), and analysis of a southern European cohort found a drop in dementia prevalence, as well (Lobo et al., 2007).
Trends seem different in Asia. One study documents a rising number of dementia and Alzheimer’s disease cases over the past two decades in Japan (Sekita et al., 2010), and a large meta-analysis found that dementia burden in China may be far larger than previously estimated (ARF related news story). Meanwhile, Yu Tzu Wu, a graduate student in Brayne’s lab, analyzed epidemiological studies of dementia in China, Hong Kong, and Taiwan (Wu et al., 2013). Wu found that apparent increases in prevalence “melted away when she took into account methodological changes over time,” Brayne said. “This highlights the need to hold the methodology steady when making inferences about change (in dementia estimates).”—Esther Landhuis
No Available Comments
One facet of neurodegeneration that continues to bewilder researchers is that the same miscreant protein can turn up in many different disorders. Take tau, for example. This protein misfolds and aggregates in more than 15 tauopathies, each with its own unique pattern of neurodegeneration and clinical symptoms. Rather than being merely a marker of neurodegeneration, as is widely accepted, could tau seed these disparate pathologies? According to work presented at the Alzheimer's Association International Conference held July 13-18 in Boston, Massachusetts, the answer may well be yes. Marc Diamond, from Washington University, St. Louis, Missouri, reported that tau forms a multitude of strains, each with its own morphology and toxic properties. The researchers have isolated some of these strains from patient brain and passaged them in cells. The findings, which some AAIC attendees considered to be a highlight of the meeting, lend solid support to the idea that different strains of the same protein may underlie different tauopathies. If true, this would have important clinical implications, say researchers.
"These findings tell us it will be possible to describe diseases based on an ensemble of conformers," said Diamond. "That brings a level of specificity and understanding that could greatly impact diagnosis and therapy." The work also indicates that tau forms bona fide prions, according to Diamond, who does not use that term lightly.
Researchers in the field of neurodegeneration have grappled with the term prion. They generally reserve it for disorders that are infectious, including bovine spongiform encephalopathy in cows, and scrapie in sheep. Aβ, tau, α-synuclein and other aggregation-prone proteins that undergo template-directed misfolding are often loosely referred to as prion-like. However, in Boston, Diamond claimed that tau satisfied the basic cell biology properties of true prions: It forms strains that are stably passed from mother cell to daughter cell; it can be taken out of the cell and reintroduced into a naive cell where it continues to propagate; and it forms multiple conformers, or strains that do all of the above. These are all characteristics of yeast prions. "We have demonstrated stable, faithful, indefinite reproduction of tau conformers in living systems," said Diamond, adding that this has never been done before with other than traditional prions.
Scientist who attended the symposium were excited by the findings. "I think this work is just fabulous," said Lary Walker, Emory University, Atlanta, Georgia. "The idea that they can take the aggregates of one morpho-type and propagate it for generations is remarkable."
Tau Strains in Cell Culture
En route to discovering tau prions in diseased human brain, David Sanders, a post-doc in Diamond’s lab, found them in stable cell lines. He used tau fibrils made in vitro to seed aggregation of a tau fragment expressed in monoclonal cells as a yellow fluorescent protein chimera. Sanders chose to express a repeat domain (amino acids 244-372), because full-length tau suppresses cell division and he wanted to propagate the cells for as long as possible. Seeding induced aggregation of the yellow chimera. As the cells grew and multiplied over the next 20 days, Sanders saw a gradual loss of aggregates. However, a small percentage of cells retained obvious yellow inclusions even up to six months and many cell generations later, when the initial tau seed was long gone from the culture medium.
While re-plating cells that propagated these tau aggregates, the researchers noticed something intriguing—as individual colonies grew, they did not all look the same. Some had diffuse perinuclear deposits, others had a few dense inclusions away from the nucleus (see figure below).
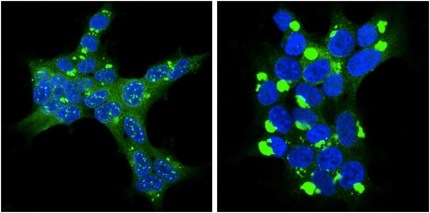
Protein Strains: Tau (green) strains clone 9 (left) and clone 10 have different morphologies. Image courtesy Marc Diamond and David Sanders
In Boston, Diamond reported that these different tau aggregates had different physical properties. On protease digestion, some completely broke apart, while others were resistant, revealing one or two bands on Western blots. The strains seem to have different toxicities, too, said Diamond in response to questions from the audience. Cells poorly tolerate one strain, dubbed clone 9. Cells carrying clone 9 quickly die if stressed, but harbor clone 10 without duress. When taken out of the cell and injected into fresh cells that had never seen tau aggregation, these strains self-propagated through daughter cells, forming identical new inclusions and retaining their characteristic biochemical properties, just like prions do.
Strains from Human Brain
Fascinating, but this could be just an in-vitro phenomenon. To test whether these strains are related to pathological tau aggregates found in the human brain, Sanders seeded aggregation using brain tissue samples taken from people who had had Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, Pick's disease, or other tauopathies. At AAIC, Diamond showed that these samples seeded tau aggregates with different morphologies. The inclusions varied from ordered to disordered, from speckled to mosaic, and each seemed to have different propensity to cause cell death, said Diamond. Samples from AD brain tissue all generated cell colonies with speckled inclusions (see image below), except for samples from one patient.
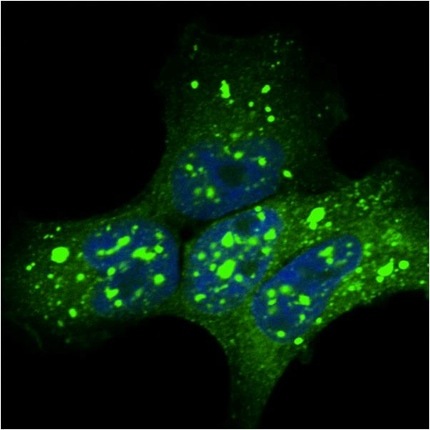
An Alzheimer’s disease tau strain: Tissue samples from AD brain propagated tau strains (green) in monoclonal cells Image courtesy Marc Diamond and David Sanders
Diamond's talk generated a fusillade of audience questions. What is the nature of the speckles? Do the infected cells survive because they have developed compensatory mechanisms? Does one strain dominate over the other? On the latter, Diamond said that clone 9 dominates over clone 10, but he has yet to to characterize the inclusions with biophysical techniques. Diamond is also studying RNA expression in the cells to see if different tau strains elicit different responses.
The findings suggest that the established concept of explaining different tauopathies by the location in the brain where the pathology first emerges may no longer be adequate, Diamond told Alzforum. He believes that location and strain are both important characteristics, as is the mechanism by which tau strains infect cells.
One consequence of this work could be that doctors better characterize, diagnose, and treat tauopathies. Diamond envisages being able to test human tissue samples, perhaps cerebrospinal fluid, or even blood, for tau strains. "We are working on a way to detect these strains by conformation," he told Alzforum. He believes that if he can correlate disease with a particular mix of strains, this could replace standard neuropathological diagnosis and also help development of treatments. Over the long term, Diamond sees therapy, particularly with antibodies, as being strain-specific. In the short term, he said strain-based diagnosis would be a huge help in designing and interpreting clinical trials, which have so far yielded no new drugs for AD or other tauopathies.—Tom Fagan.
No Available References
No Available Further Reading
Ligands that light up tau on brain scans generated plenty of buzz among researchers attending the Alzheimer's Association International Conference, held July 13-18 in Boston. Presenters at the meeting offered the latest data on three compound classes. They are T807 and T808, being developed by Eli Lilly and Company through their subsidiary Avid Radiopharmaceuticals. Then there is the THK serious of quinoline-derived compounds named after Tohoku University in Sendai, Japan. The third, and newest, player is PBB3, a benzothiazole derivative similar to Pittsburgh compound B (PiB). PBB3 is being developed at National Institute of Radiological Sciences in Chiba, Japan.
The number of people who have undergone positron emission tomography (PET) with these tracers is still small—in the dozens thus far. That said, early human data show two promising, crucial characteristics, said William Klunk of the University of Pittsburgh in Pennsylvania. Several of the compounds generate a good signal-to-noise ratio, and they label brain areas where tau pathology occurs. One important step to validate the ligands, said Klunk, will be to formally prove they highlight the same areas of the brain where tau aggregates are found on postmortem examination. Lilly presented early postmortem results from one study participant who died of unrelated causes soon after having a T808 scan.
In addition to postmortem validation, researchers have to answer plenty of questions about these tau ligands before they are ready for prime time. Will the ligands help identify non-AD tauopathies, such as chronic traumatic encephalopathy (CTE), corticobasal degeneration (CBD), or progressive supranuclear palsy (PSP)? Since aggregating tau takes different forms, one cannot assume that one ligand will bind them all, noted William Jagust, University of California in Berkeley, and other PET experts who spoke with Alzforum. One crucial test, Klunk noted, is that the ligands must bind tau, but not Aβ amyloid. This is difficult because the latter is much more abundant in the AD brain and could cause false positive scans unless the ligand has a much higher specific affinity for tau than for Abeta. Nonspecific binding plagued early tau tracers such as FDDNP, which are said to bind both beta amyloid and tau deposits (Small et al., 2013).
Lilly’s Compounds Offer Initial Post-Mortem Data
Lilly acquired the fluorine-tagged ligands T807 and T808 from Siemens Molecular Imaging Biomarker Research in April 2013 (see Alzforum News story). These compounds came out of compound library screens that used human brain slices laden with tau tangles as binding targets. T807/8 have different heteroaromatic core structures, but bind the same pocket of tau with similar affinity (Zhang et al., 2012) .
Thus far, Siemens and Avid researchers have run T807 PET scans of 10 people—three with a low probability of AD and seven with a high probability, based on clinical examination. In his AAIC presentation, Mark Mintun from Avid expanded on previously published data from six of those people (see ARF related news story). The strength of T807 labeling correlated with the severity of cognitive defects experienced by the volunteers, as measured by the Mini-Mental State Exam (MMSE), said Mintun.
One outlier broke the trend. The researchers originally diagnosed this participant with AD, but the scan suggested very little tau pathology. Later, the researchers scanned this same person for Aβ pathology, and that, too, was negative. Mintun concluded the person likely never had AD (see image below).

PET imaging with T807: Across normal people and those with likely AD (red box), signal strength increased with worsening scores on the MMSE. One outlier (yellow box) turned out to be amyloid negative. Image courtesy of Mark Mintun, Avid Radiopharmaceuticals, Philadelphia, Pennsylvania.
Mintun concluded that T807 entered the brain and labeled traditional tau-filled regions, including the temporal, parietal, and occipital lobes. The next steps, he said, will be to characterize how those labeled regions correlate with the types of cognitive decline experienced by participants. Hartmuth Kolb, formerly at Siemens but now also at Avid Radiopharmaceuticals, tested T808 in 12 people, eight with cognitive impairment (MCI or AD) and four healthy controls. As with T807, Kolb observed that the worse off a participant was cognitively, the more tau the tracer stained in their brains, with labeling in the frontal, temporal and parietal lobes as well as the hippocampus.
Furthermore, Kolb presented preliminary data on postmortem validation for T808. One 87-year-old man in the study, who had a diagnosis of likely AD, died of a pulmonary embolism two weeks following his scan; he had agreed to donate his brain. Kolb and colleagues stained his brain with the AT100 antibody for phosphorylated tau. They saw that the post-mortem immunostaining of the frontal, temporal and parietal lobes and the hippocampus agreed with the regions lit up by the PET ligand, Kolb said.
Avid has started investigations to determine if these ligands label tauopathies other than AD. The researchers are checking post-mortem brain samples from several tauopathies with the fluorescent marker T557, which has a similar structure to T808 and likely binds the same kinds of aggregated tau. This label lit up cases of Pick’s disease, CTE and frontotemporal dementia. “We should be able to see tau in these brains” with T808, Kolb suggested.
Beyond Kolb and Mintun, PET experts at different academic institutions are beginning to study these tracers. In years past, Avid Radiopharmaceuticals had readily shared AV45/florbetapir with the research community, in effect tapping the field’s collective expertise to study the compound and gather data that helped move it through development quickly.
THK Team Reports Improved Specificity
For their part, research collaborators from Japan and Australia came to Boston with news of the fluorine-tagged THK tracers, including first-generation THK-523 and second-generation THK-5105 and THK-5117. THK-523 also came out of a screen for tau ligands, which identified several quinoline- and benzimidazole-based compounds (Okamura et al., 2005 ). In mice, THK-523 has good affinity for tau fibrils, but not amyloid (Fodero-Tavoletti et al., 2010). Nobuyuki Okamura, of Tohoku University, reported on 20 human subjects. Ten AD patients and 10 healthy controls underwent scans with THK-523. It labeled areas known to accumulate tau paired helical fragments (PHFs), including the temporal, parietal, and orbitofrontal lobes, and the hippocampus of people with AD.
However, THK-523 binds too strongly in white matter, making it hard to differentiate AD and control profiles, said collaborator Victor Villemagne of Austin Health in Melbourne, Australia, in a discussion with Alzforum. Instead, the researchers are now pursuing the new compounds THK-5105 and THK-5117. These share similar structures with 523 but are optimized for higher affinity binding to tau aggregates (Okamura et al., 2013). THK-5105 shows no retention in white matter, making it better suited to distinguish cases and controls, Villemagne said.
Okamura reported on eight people with AD and eight healthy controls who underwent THK-5105 imaging. The labeled areas correlated with known distributions of tau in the AD brain—the hippocampus and neocortex, in particular the inferior temporal cortex—but not with areas where the PiB amyloid tracer bound. As with other ligands, the 5105 binding increased with the severity of the dementia based on MMSE scores. Okamura told Alzforum that preliminary data on THK-5117 indicates it has better pharmacokinetics than THK-5105; however, the researchers will determine which is best after these first studies are complete.
Since the THK compounds bind PHFs, Villemagne said he suspects they will interact with tau not only in AD, but also in Down syndrome and CTE. THK-5105, but not THK-523, showed some interaction with corticobasal lesions, he said. The compounds are insensitive to Pick bodies and the tangles in PSP, Okamura said. Other clinician-scientists expressed great interest in using PET tracers to better understand the different conformations by which tau aggregates and deposits in the different tau diseases. At this point, no researcher interviewed by Alzforum anticipates that there will be a single pan-tau PET tracer for all tauopathies.
Okamura told Alzforum he hopes to perform longitudinal studies of tau tracers in a large group of people with AD, MCI, and preclinical AD. “By the assessment of spatial and temporal pattern of tau deposition, we will understand the role of PHF-tau in AD progression, cognitive decline, and neurodegeneration,” he suggested.
Progress with PBB3
Finally, researchers led by Hitoshi Shimada of Japan’s National Institute of Radiological Sciences have tested PBB3 in 10 healthy controls, five people with MCI, and 13 with AD. PBB3 labeled large areas of the brain, including the medial temporal cortex, hippocampal limbic system, and neocortex. The tracer’s signal was highest and most widespread in the most severe AD cases. It showed no overlap with amyloid beta ligand binding, Shimada claimed. Tau imaging may reflect the severity of cognitive defects better than amyloid scans, Shimada suggested. He declined to say what specific tauopathies PBB3 might label.
PBB3 is structurally similar to PiB, but has a molecular spacer in its middle that makes it bind to tau. Some scientists who spoke with Alzforum cautioned that they would like to see further data clarifying how well PBB3 distinguishes tau from Abeta amyloid. Others noted that in its current form PBB3 might be less widely useful than the other compounds because it relies on carbon-11, which has a much shorter half life than fluorine-18. Users must have an on-site cyclotron for carbon-11 imaging, making the label less practical.
Evolving Imaging
The AD field has long awaited a validated tau tracer. The advent of promising ligands “for the second major molecular pathology of the disease is a very exciting development,” commented Andrew Saykin of Indiana University in Indianapolis. However, he noted that the wait goes on, as the current crop of tracers requires more validation before regular use in the clinic, or even use to follow people’s tau burden in clinical trials.
Validated tau tracers could help researchers answer many questions. “They will clearly define the relative time course of amyloid deposition versus tau,” said Christopher Rowe of Austin Health in Melbourne, Australia. Rowe’s site is testing the THK compounds. Based on tau in cerebrospinal fluid, scientists estimated that tau changes after amyloid, but imaging would confirm whether the same sequence occurs in the brain.
In clinical trials, tau tracers could help scientists identify the right subjects for potential tau interventions, and follow their progress. “This is becoming a much more urgent need because tau is being recognized more as a potential target for new therapeutics. We need a tool to evaluate these drugs,” commented Ralph Nixon of the Nathan Kline Institute in Orangeburg, New York, who moderated a press conference about the topic. Tau PET scans might also help doctors decide when a person should start a future anti-tau therapy.
Eventually, clinicians might be able to use tau imaging to differentiate Alzheimer’s from normal aging or other conditions, researchers suggested. Tau correlates with disease symptoms better than does amyloid, and the latter can also show up in what appears to be normal aging, noted Jagust. Tau starts to show up as cognitive decline becomes imminent, Rowe said, and thus might help doctors predict symptoms better. However, scientists who spoke with Alzforum noted that the challenges in getting amyloid imaging reimbursed by insurers (see ARF related news story) do not bode well for regular clinical use of tau imaging.
The low abundance of tau aggregates in the brain, and its multiple forms, make tracing this protein a much more complicated task than it was for Aβ, said Klunk. Nonetheless, Rowe noted, the field has seen “an explosion of research with tau imaging.” He anticipates lots more news at next year’s AAIC in Copenhagen, Denmark.—Amber Dance.
No Available Comments
No Available Further Reading
A creepy idea is taking hold in Alzheimer’s research. Like a computer worm that travels through networks corrupting perfectly good files, toxic forms of tau bore from neuron to neuron turning good protein into bad along the way. Scientists at the Alzheimer’s Association International Conference (AAIC), held July 13-18 in Boston, discussed how aggregated tau makes this jump from neuron to neuron, how that affects the invaded cells, and how medicines might stop the process in its tracks. The upshot was that although researchers have come to associate this cell-to-cell transmission with misfolded tau, normal tau might travel the same way. Moreover, new data suggest that misfolded tau does not kill neurons directly or cut them off from their neural networks. Despite many gaps in current understanding, the transport of misfolded tau undoubtedly promotes dementia, leading many companies to pursue new therapeutics in the form of antibodies that grab onto the bad tau before it spreads.
Tau neurofibrillary tangle pathology as visualized at autopsy follows a distinct progression pathway in the AD brain. Starting in the locus coeruleus, it moves toward the transentorhinal cortex, hippocampus, and beyond. Importantly, it does not merely diffuse out radially from its point of origin; rather, it travels along neural circuits (Braak and Del Tredici, 2011). A similar spread occurs in animal models. For example, in mice that express mutant tau only in the entorhinal cortex, tau pathology travels to connected regions in the brain as the animals age (see Alzforum related news story). Researchers have come to believe that tau spreads like a prion, with pathological protein converting native protein to a misfolded, pathogenic form (see Alzforum related news story).
Cell-to-Cell Transmission—Not Just For Toxic Tau
Some researchers at AAIC turned their attention to how ordinary, non-mutant tau might move throughout the brain. Amy Pooler of King's College London, U.K., presented evidence that transport of wild-type tau among cells is a normal phenomenon. She also claimed it is induced by neuronal activity. In a paper that took the Alzheimer’s field by surprise, Pooler had reported previously that pathogenic tau hijacks this physiological transport process to move from cell to cell (see Braak et al., 2011). Specifically, Pooler found that cultured neurons stimulated with potassium chloride or glutamate released tau into the cell medium. The agonist (S)-AMPA, which stimulates AMPA glutamate receptors, had a similar effect. On the other hand, quenching neuronal activity with tetrodotoxin, or blocking synaptic vesicle release with tetanus toxin, reduced the release of tau in response to (S)-AMPA.
Pooler’s work convincingly shows that neuronal activity drives tau out of cultured neurons, commented David Holtzman of the Washington University School of Medicine in St. Louis, Missouri, in an email to Alzforum. Still to be determined, he added, are whether the same occurs in vivo and what the mechanism of tau release might be. Notably, active neurons also release amyloid-beta (see Alzforum related news story).
“Tau release may be a normal process in the healthy brain,” concluded Pooler in an email to Alzforum. Other evidence supports this idea, she added. Tau flows in the interstitial fluid of healthy mouse brains (see Alzforum related news story) and there is evidence that extracellular tau stimulates neurotransmitter receptors (Gómez-Ramos et al., 2008). The process of tau transfer could offer new potential drug targets, commented Benjamin Wolozin of Boston University.
Marc Diamond, also from Washington University, agreed. His group recently reported that once tau escapes into the extracellular space it can bind to heparan sulfate proteoglycans (HSPGs) on the surface of neighboring cells (see Holmes et al., 2013). Blocking HSPGs prevented tau transmission to other cells. HSPGs appear to act like a receptor for tau and also for α-synuclein, or by knocking out Ext1, an enzyme essential for HSPG synthesis. A suite of enzymes are known that modify HSPGs and turn them into tau receptors, noted Diamond. "These are all enzymes with active sites—potentially druggable," he told Alzforum. "This suggests an entirely new class of drug targets."
Modeling Sporadic Disease
Luc Buée of Inserm in Lille, France, has for years studied normal tau, in his case the wild-type protein that aggregates in the brains of people with sporadic tauopathies. Buée noted that a classic model for tau transmission, in which injected tau seeds spread from one part of the brain to others, relies on the delivery of mutant tau into mice primed to spread that pathology by their overexpression of human, wild-type tau. Classic Abeta seeding models work similarly (see Alzforum related news story; also Clavaguera et al., 2013). At AAIC in Boston, Buée described a similar model, with one crucial difference—it relies on only wild-type tau (see Alzforum related news story). Buée and colleagues used a lentivirus to deliver the wild-type human tau gene into the hippocampus of normal rats. Neurodegeneration and tangle-like structures spread from the site of injection along neuroanatomical pathways that connect networked parts of the brain. “Using wild-type tau, we were able to induce a type of pathology,” Buée concluded in an interview with Alzforum. This model could be a closer match for sporadic human disease, in which only wild-type tau is present.
To better understand the spread of tau from the viral injection site, Buée and colleagues added to the lentiviral vector a piece of DNA coding for V5. This peptide is widely used to tag and study the fate of proteins in the cell. That way, the researchers could distinguish the tau encoded by the injected gene from the endogenous version. To the researchers’ surprise, they observed that this V5-tagged tau spread all the way from the hippocampus to the olfactory bulb, traveling anterogradely along projection pathways. Buée told Alzforum that he believes tau undergoes some sort of active transfer from neuron to neuron. Like Pooler, he thinks tau transport is a normal process, and that when additional stressors turn tau toxic, its pathogenic kin follows the same trails.
Counterintuitively, the pathogenic tau may travel at a more leisurely pace. When the researchers expressed proline-301-leucine tau mutant, which causes neurodegeneration, they saw that deposits spread more slowly than the wild type. The researchers speculated that mutant tau does not spread so fast or so far because it rapidly aggregates, getting stuck in cells near the injection site. “This finding is particularly interesting. It suggests that it is the soluble form of tau that is propagated and that tangle formation might prevent the release of tau into extracellular space,” agreed Pooler. This does not preclude the role of mutations in aggressive tauopathies, since in that case all neurons have the mutant tau and it can cause damage without spreading, Buée noted. This new rat model of progressive tauopathy may be useful to investigate how tangles form, and also to test potential therapeutic compounds that may slow tau aggregation, Pooler added.
Full of Tangles, But Ready to Work
One transported, toxic tau forms intracellular tangles. Are these toxic waste dumps contributing to disease, or relatively safe places to store undesirable types of tau? Work presented by Susanne Wegmann, of Bradley Hyman’s group at Massachusetts General Hospital in Charlestown, suggests the latter. At AAIC, Wegman reported that tangled tau barely disrupts neural function or brain networks. The work was also reported by Wegmann’s colleague Kishore Kuchibhotla at the International Conference on Alzheimer’s and Parkinson’s Diseases in March (see Alzforum related news story).
Wegmann set out to directly compare tangle-laden neurons to neighboring, tangle-free nerve cells. She studied transgenic mice that overexpress human P301L tau and develop neurofibrillary tangles in their cortex. To assess individual neuron function, she used moving gray bars as visual stimuli to activate neurons in the visual cortex. As output, she recorded calcium flares in those neurons by in-vivo confocal microscopy and a calcium sensor called Yellow-Cameleon 3.6. Following the stimulation experiments, Wegmann sacrificed the mice and used immunostaining to identify neurons that contained, or lacked, tangles. She observed no difference in the responses of tangle-loaded neurons and their tangle-free neighbors.
“Even though these mice carry a high tangle load, they have no major neuronal network dysfunction in the visual cortex,” Wegmann concluded. She suggested that tangles themselves may not be directly neurotoxic. “These data…might even lend support to the idea that sequestration of tau into these tangles might be a protective mechanism,” added Pooler.
These findings have potentially good and bad implications, Buée told Alzforum. On the upside, it suggests that neurons with tangles in them remain functional and wired into the brain’s networks. If a treatment could clear away the toxic forms of tau, these neurons could be saved. However, since tangle-filled neurons remain connected, they likely could transmit their pathology to others in the network, further spreading the disease, said Buée. Other scientists interpreted Wegman’s finding as raising the notion that tangle-busting drugs could unleash more toxic forms of tau from a relatively less toxic storage compartment – a discussion that echoes from years past when plaque-busting drugs were being debated.
Modeling and Targeting Tau Transmission
If toxic tau travels, then a roadblock could halt disease, many researchers have reasoned. Nearly a dozen companies have turned their attention to immunotherapy using antibodies that bind tau and prevent its trans-synaptic spread, said Michael Hutton of Eli Lilly and Company in Windlesham, UK. Lily is among the companies pursuing this approach.
Those companies need ways to test their therapies. Zeshan Ahmed of Lilly’s Surrey, U.K. site was looking for a system that uses tau injection but is faster and more suitable for preclinical study of drug candidates. As with earlier models, Ahmed used brain extracts from old proline-301-serine-tau mice as the source of tau. But instead of targeting mice that over-express wild-type human tau, he injected it into young P301S mice. Already expressing mutant tau, these young mice were primed for tau transmission. Indeed, they demonstrated a “startling induction of tau pathology” within just 2 months, Ahmed said. Researchers have performed similar experiments injecting extracts from the brains of people with taopathies into P301S mice (see Alzforum related news story).
Mild tau pathology appeared just two weeks after injecting the brain extracts into the hippocampus. By one month, the damage had spread to the opposite side of the brain from the injection, and this contralateral pathology was more severe by two months. Again, the tau moved along neuroanatomical pathways instead of merely diffusing to neighboring regions. “It is a clear demonstration that the pathology spreads to regions that are synaptically connected,” said Hutton. “You have the tau pathology jumping across the synapses into a new brain area.” Despite this pathology, neurons remained present and alive.
“I like the speed and ease of this model,” said Wolozin. “The current models take a long time to see a bit of tau transfer.” The model will be useful for testing drugs as well as to understand tau transport biology, he suggested. The P301S strain could be crossed with knockouts for any number of genes suspected of involvement in tau transfer, and checked for effects on the post-injection pathology. The high level of tau aggregates also makes the mice suitable for biochemical studies, Wolozin said.
Hutton said the tau horizon is looking brighter than in earlier days, when efforts to affect tau phosphorylation and aggregation fizzled in human trials. Antibodies to block transfer are “currently the hottest area in tau therapeutics,” Hutton said. Lilly has already had some success reducing tauopathy in mice (Chai et al., 2011). Hutton predicts several of these therapies will move into the clinic over the next couple of years.—Amber Dance
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.