A Sinister Side to α-Synuclein—Blocking Mitochondrial Protein Import
Quick Links
Mitochondria in dopaminergic neurons go haywire in Parkinson’s disease, ramping up oxidative stress and eventually killing the cells. New evidence puts α-synuclein at the scene of the crime. In the June 8 Science Translational Medicine, researchers led by Timothy Greenamyre at the University of Pittsburgh report that oligomeric forms of α-synuclein bind the mitochondrial receptor TOM20. In rat and culture models, this interaction gummed up protein import into the organelle, eventually causing cellular respiration to flag and reactive oxygen species to rise. Knockdown of α-synuclein, or overexpression of TOM20, prevented mitochondrial problems. Importantly, the authors found α-synuclein and TOM20 bound together in tissue from Parkinson’s brains as well, suggesting the same process occurs in human disease. “Now we know α-synuclein damages mitochondria through a specific mechanism. That raises the possibility we could target it therapeutically,” Greenamyre told Alzforum.
Other researchers appreciated the insight the data provide into basic disease mechanisms of Parkinson’s. “This identifies another key target for α-synuclein that falls in with a set of pathways genetically linked to the disease, and opens up new avenues for investigation,” said Benjamin Wolozin at Boston University. Julie Andersen at the Buck Institute for Research on Aging, Novato, California, noted how this addresses unanswered questions. “Previous studies have linked increases in α-synuclein to mitochondrial dysfunction, but the mechanisms has been elusive until now.”
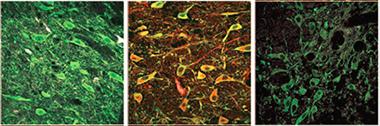
Mitochondrial Attack.
Red fluorescent proximity ligation assays reveal that in tyrosine hydroxylase-positive (green) dopaminergic cells (left), rotenone causes α-synuclein and the mitochondrial protein TOM20 to bind (middle); knockdown of α -synuclein abolishes this interaction (right). [Courtesy of Science Translational Medicine/AAAS.]
Much of the previous research on α-synuclein has focused on its effects on autophagy and vesicle recycling (see Jun 2012 news; Apr 2013 news; Feb 2014 news). Meanwhile, a parallel line of research linked Parkinson’s risk genes such as parkin and PINK1 to dysfunction and improper disposal of mitochondria (see Mar 2014 news; Oct 2014 news; Sep 2015 news). Some studies tied the two together, reporting an association between excess α-synuclein and mitochondrial stress, but it was not clear which came first, or what the mechanisms might be (see Hsu et al., 2000; Betarbet et al., 2006; Zaltieri et al., 2015).
Greenamyre was intrigued by the fact that when α-synuclein interacts with membranes, it can assume a helical shape (see Davidson et al., 1998). He noted this shape resembles the structure of the mitochondrial targeting signal (MTS) found on proteins destined for the organelle. Proteins containing an MTS tag bind TOM20 and its partner TOM22, triggering import into mitochondria. Greenamyre wondered if α-synuclein might bind TOM20 as well.
To test this idea, joint first authors Roberto Di Maio and Paul Barrett examined dopaminergic neurons from the brains of two rat Parkinson’s models, one produced by administering the poison rotenone, the other by viral overexpression of α-synuclein in the substantia nigra. The authors used a proximity ligation assay to detect the interaction of α-synuclein and TOM20. In this technique, antibodies against the two proteins of interest carry probes that hybridize when in close contact to generate a fluorescent signal. In both PD models, the authors saw an eightfold surge of α-synuclein and TOM20 binding over control levels. At the same time, levels of Ndufs3, a mitochondrial complex I protein, dropped, suggesting a disruption in the normal flow of proteins into mitochondria. Knocking down α-synuclein in rotenone-treated rats by about one-third abolished the α-synuclein-TOM20 interaction and restored normal mitochondrial import (see image above).
What forms of α-synuclein cause problems? To answer this, the authors turned to in vitro assays using purified, recombinant α-synuclein that they modified using various protocols. In isolated mitochondria and in dopaminergic cell cultures, monomeric, nitrated, or fibrillar α-synuclein did not bind TOM20 and had no effect on mitochondrial import. On the other hand, α-synuclein that had been oligomerized in vitro, phosphorylated at site S129, or exposed to dopamine glommed onto TOM20 and cut mitochondrial import in half. After exposure to these forms of α-synuclein that blocked protein import, TOM20 ceased to bind its partner TOM22, as seen in another proximity-ligation assay. When TOM20-TOM22 binding failed, mitochondrial respiration dropped by one-third, and protein oxidation in mitochondria doubled.
The authors wondered what the toxic species of α-synuclein had in common. They did not see any obvious differences in secondary structure between toxic and non-toxic species. Except for the fibrillar form, all appeared to exist in a largely unfolded state, with only small amounts of helical structure. However, in all three preparations of the toxic species, about one-quarter to one-third of the synuclein was trimers and tetramers, as judged by size on non-denaturing SDS gels. Non-toxic preparations, on the other hand, comprised mostly monomers, dimers, and high-molecular-weight material. Small oligomers appear to be the culprits in mitochondrial dysfunction, the authors concluded.
The authors believe these toxic species are unlikely to be the physiological tetramers described by Dennis Selkoe at Brigham and Women’s Hospital in Boston and others (see Apr 2015 conference news). Unlike those labile forms, the toxic variety are highly stable, even resisting boiling, Greenamyre told Alzforum. He plans to analyze their structure further, and is also examining how genetic variants of α-synuclein that cause PD interact with mitochondria, and what forms they assume.
How relevant are the findings to human disease? The authors examined postmortem sections from the substantia nigra of five PD brains and four controls. As in the animal models, they found about eightfold more α-synuclein-TOM20 complexes using the proximity ligation assay, and 50 percent less mitochondrial Ndufs3, as assessed by immunocytochemistry, in the PD brains. Greenamyre is currently characterizing which species of α-synuclein bind TOM20 in these brain samples.
The identification of this mitochondrial mechanism suggests new therapeutic strategies, Greenamyre believes. In dopaminergic cell cultures treated with toxic forms of α-synuclein, a fourfold overexpression of TOM20 prevented mitochondrial import problems and maintained cellular respiration. Surprisingly, expressing just an MTS sequence also protected cells from α-synuclein. Because MTS binds TOM20, the authors expected it might also poison mitochondria. Instead, the peptide appeared to block α-synuclein binding to TOM20, while not interfering with the TOM20-TOM22 interaction. The reason is not clear, but possibly MTS jumps quickly on and off TOM20, in contrast to the more stable binding of α-synuclein, Greenamyre speculated. He is currently testing these treatment strategies in animal models and looking for improvements in pathology and behavior.
The interaction between α-synuclein and mitochondria may provide clues as to why dopaminergic neurons in particular fail in PD, Greenamyre noted. In control rats, these neurons gave off only a weak TOM20-TOM22 interaction signal compared to other neurons, suggesting their mitochondrial import machinery might be feeble and easily perturbed. Synuclein toxicity, such as that caused by the abundant dopamine in these cells, might be the culprit, Greenamyre hypothesized (see May 2002 news). To test this, the authors knocked down α-synuclein in the substantia nigra of wild-type rats. The TOM20-TOM22 interaction picked up, and protein oxidation dropped by about half, suggesting rescue of mitochondrial function. Wolozin found the data intriguing. “This may answer the age-old question: What is it about dopaminergic neurons that renders them most vulnerable to Parkinson’s disease?” he said.—Madolyn Bowman Rogers
References
News Citations
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- Synuclein Resists and Disrupts Autophagy
- LRRK2 Interactions Identify New Parkinson’s Genes, Implicate Autophagy
- New Role for PINK1 Offers Clues about Parkinson's Pathology
- ALS, Parkinson’s Proteins Co-Mingle in Mitochondria Destruction Pathway
- PINK1 Can Act Alone to Destroy Mitochondria, But Parkin Helps
- Form and Function: What Makes α-Synuclein Toxic?
- Dopamine Renders α-Synuclein Toxic to Neurons
Paper Citations
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000 Aug;157(2):401-10. PubMed.
- Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006 May;22(2):404-20. PubMed.
- Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A. Mitochondrial Dysfunction and α-Synuclein Synaptic Pathology in Parkinson's Disease: Who's on First?. Parkinsons Dis. 2015;2015:108029. Epub 2015 Mar 31 PubMed.
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998 Apr 17;273(16):9443-9. PubMed.
Further Reading
News
- Parkin Loss AIMPs Up Dopamine Neuron Death in Parkinson's
- Shape of α-Synuclein Aggregates Influences Pathology
- Electron Microscope Yields Finer Structure of α-Synuclein, Aβ Fibrils
- Potential Parkinson’s Treatments Target α-Synuclein, Cell Replacement
- Gene Captain, PGC-1α, Abandons Mitochondria in PD
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
Primary Papers
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. 2016 Jun 8;8(342):342ra78. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
U.K. Dementia Research Institute at University College London
Harvard Medical School, Brigham and Women’s Hospital
The study by Di Maio et al. is an important contribution to our understanding of the role of mitochondria and α-synuclein (α-syn) in the pathogenesis of Parkinson’s disease. The authors report here an aberrant interaction of certain modified or wild-type forms of α-syn with impaired mitochondrial protein import, giving us potential insight into two converging pathways that have been associated with early PD pathogenesis: α-syn modifications/misfolding and mitochondrial dysfunction. In a series of elegant experiments, mainly using a technique that allows probing for protein-protein interaction in ex vivo samples, the proximity ligation assay (Söderberg et al., 2006), the authors establish a robust interaction between α-syn and TOM20 that seems to correlate with pathology and lead to toxicity. Furthermore the authors are able to give us pointers toward one of the oldest questions in PD: What is it about dopaminergic neurons in the substantia nigra (SN), among all α-syn-expressing cells, that makes them particularly susceptible to α-syn mediated toxicity? Earlier work by the Surmeier group (Guzman et al., 2010) pointed toward the particular pace-making activity of SN neurons that leads to increased calcium flux and mitochondrial oxidative stress, something that, interestingly, recently has been linked to α-syn-mediated toxicity (Luth et al., 2014). The second, entirely novel connection presented here again converges at the mitochondria: Low baseline interaction between TOM20 and its co-receptor TOM22, which makes mitochondria especially susceptible to the disruptive effect of pathological α-syn species, seems to be a hallmark of dopaminergic neurons. In the future, strengthening the interaction of both TOM transporters or lowering the concentration of pathological α-syn could be promising therapeutic routes that need to be explored.
An important question remains, though: What forms of α-syn mediate this toxic interaction? It has been a challenge for the field to address the question of what exactly the different pathology-associated α-syn species are. While the authors established the aberrant interaction of oligomeric, dopamine-modified, and S129E (phosphomimetic) α-syn (but not nitrosylated or fibrillar α-syn) with mitochondria in patient tissue, what these variants have in common remains elusive. The authors speculate that trimeric or tetrameric structures may be important (at least for dopamine-modified and S129E, which are found as defined species and should be part of the “smear of different species” in the oligomeric preparation). It seems unlikely that these species are the same physiological tetramer described by us (Bartels et al., 2011) and others (Burré et al., 2014), given that the former have significantly less secondary structure than the physiological tetramer. Oligomers, dopamine-modified, and S129E variants are characterized as unfolded by the authors, which leaves open the possibility that these interact with the mitochondrial membrane and form helical species just as the monomer does, pointing toward a normal physiological membrane interaction of α-syn that is modified in the disease condition, as described before (Kamp et al., 2010). Given that the authors describe a relatively strong and specific interaction of α-syn with TOM20, it should be possible to identify a specific structural determinant in the α-syn species that mediates the interaction, and it will be interesting to see if the authors (or another group) can provide that in subsequent studies.
A caveat is the mode of delivering purified recombinant α-syn to cultured cells. Given α-syn’s conformational flexibility and strong context-dependent structure, this approach is fraught with peril. While the synthetic forms of α-syn in model systems do recapitulate the toxicity seen in vivo in α-syn-overexpression systems, it is not clear that this extends to the α-syn-TOM20 interaction seen in patient brain. Further characterization of the synthetic species that mediate toxicity in vitro and finding common properties with TOM-20 associated α-syn species in patient samples would be necessary to specifically target these species in disease. But maybe that is not even necessary, as the authors correctly state that ”it appears that strategies aimed at a general reduction of α-synuclein, rather than targeting specific modifications or aggregation states, may be most efficacious.” Thus, the most important new insight and potential new avenue toward PD disease-modifying drugs might be that stabilizing TOM20-TOM22 interactions and/or upregulating TOM20 expression could counter the detrimental effects of toxic α-syn species on mitochondria.
References:
Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006 Dec;3(12):995-1000. PubMed.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010 Dec 2;468(7324):696-700. PubMed.
Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem. 2014 Aug 1;289(31):21490-507. Epub 2014 Jun 18 PubMed.
Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011 Aug 14;477(7362):107-10. PubMed.
Burré J, Sharma M, Südhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014 Oct 7;111(40):E4274-83. Epub 2014 Sep 22 PubMed.
Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010 Oct 20;29(20):3571-89. PubMed.
University of Pittsburgh
We appreciate the comments by Tim Bartels and Ulf Dettmer, and we agree with everything they have said. As noted in our paper, and as Bartels and Dettmer reiterate, it will be very important to define the structural characteristics and commonalities of the species of α-synuclein that exert mitochondrial toxicity. Studies attempting to do so are underway.
Bartels and Dettmer also point out that there are caveats associated with delivering purified recombinant proteins to cells, a point with which we agree. However, one major advantage of our approach is that it does not rely on overexpression, which may introduce artifactual protein interactions. In fact, the use of extrinsic α-synuclein did not measurably alter the levels of α-synuclein in cells. Additionally, our protocol allowed us to control, to a large extent, the specific species of post-translationally modified α-synuclein under study.
A key outstanding question is: What species of α-synuclein are interacting with TOM20 in neurons in the parkinsonian brain? Given that only a tiny fraction of α-synuclein in a cell interacts with TOM20, and α-synuclein is an abundant protein, and TOM20 is an intrinsic membrane protein, conventional co-immunoprecipitation approaches are unlikely to be enlightening. On the other hand, as new α-synuclein antibodies are developed and validated, it may be possible to identify the toxic species in situ. In this regard, we have been able to demonstrate strong proximity ligation signal between endogenous S129-phospho-α-synuclein and TOM20 in ex vivo brain sections of substantia nigra, thereby confirming what we found in vitro with recombinant S129E-α-synuclein (Di Maio, Barrett et al., unpublished). It is hoped that other validated, conformer-specific antibodies will be similarly useful. Finally, as we pointed out, and as reiterated by Bartels and Dettmer, strategies aimed at reducing overall levels of α-synuclein may be useful, regardless of which specific forms of α-synuclein are the culprits. Similarly, strategies designed to mitigate α-synuclein toxicity to mitochondria, such as increasing levels of TOM20 or expressing a mitochondrial targeting signal peptide, may be useful therapeutically.
Roberto Di Maio and Paul Barrett contributed to this comment.
Make a Comment
To make a comment you must login or register.