Protein Screen Links Mitochondrial Regulator to Alzheimer’s Disease
Quick Links
How do proteins linked to neurodegenerative disease heighten risk? One way to answer this question is by looking at the company they keep. In the May 19 Cell Reports, researchers led by Matthias Selbach at the Max Delbrück Center for Molecular Medicine in Berlin describe a mass spectrometry method for comparing the interactions of wild-type proteins to those of disease-causing variants. As a proof of concept, the authors analyzed five mutant proteins from Alzheimer’s, Parkinson’s, and Huntington’s diseases, and spinocerebellar ataxia. They turned up 125 proteins that interacted differently with wild-type and mutant bait, a few of which had been previously known. The scientists analyzed one, the mitochondrial protein LRPPRC, which preferentially bound to amyloid precursor protein (APP) harboring the Swedish mutation, and found that APPSwe suppressed LRPPRC and mitochondrial activity in cultured cells.
Other researchers said the method holds promise. “[This method] provides a general and much-needed approach to rapid functional characterization of disease-related genes emerging from GWAS and exome-sequencing studies,” Bruce Yankner at Harvard Medical School wrote to Alzforum. Putative interacting proteins will need to be validated in model systems to ascertain their disease relevance, he added.
Many groups are building interaction maps for risk genes. These can illustrate the cellular processes most affected by a given condition (see May 2013 webinar; Sep 2013 news; Jan 2014 news). The maps tend to be constructed from protein-protein interaction databases, which are largely based on the binding of wild-type proteins (see Rual et al., 2005; Ewing et al., 2007; Szklarczyk et al., 2011). Traditional methods for examining protein interactions, such as mass spectrometry and two-hybrid screens, do not distinguish small differences in binding strength between wild-type and mutant proteins, the authors note.
To tackle this problem, Selbach and colleagues first catalogued the interactions of wild-type and disease-causing variants separately. They chose five genes: APP and PS1 for Alzheimer’s, Park2 for Parkinson’s, huntingtin for Huntington’s disease, and ataxin-1 for spinocerebellar ataxia type 1. For each, they analyzed the wild-type plus one or two variants, for 12 total test proteins. First author Fabian Hosp, now at the Max Planck Institute of Biochemistry, Martinsried, Germany, expressed each protein along with a c-myc tag in cultured cells, and purified the lysate with beads containing anti-c-myc to exclude nonspecific interactions with c-myc alone. Next came analysis of the bound material by mass spectrometry to identify proteins that co-purified with the tagged protein.
Altogether, the authors found 373 proteins that interacted specifically with at least one of their 12 exemplars. Most of these bound a particular protein variant, although some, including about 10 involved in proteolysis, bound most of the test proteins. Some known interactions turned up, such as between VCP and huntingtin, but most were new.

Mutations Change Binding. In head-to-head comparisons, some interacting proteins bound better to the wild-type version of proteins (blue), and some to mutant versions (red). The majority of interactors showed no preference (gray). [Image courtesy of Cell Reports, Hosp et al.]
Do mutated and wild-type proteins bind differently to the same partner? To answer this, the authors needed a quantitative method. They compared the interactions of normal and disease variants by expressing the tagged genes in cells labeled with either heavy or light amino acid isotopes, respectively, then purified the proteins and analyzed bound material by mass spec. The ratio of captured heavy to light forms revealed whether a protein bound preferentially to the wild-type or mutant form. One hundred and twenty-five proteins bound preferentially. Ten proteins bound better to wild-type PS1 than mutant, while two bound better to wild-type APP and two preferred APPSwe (see image above). Only a few of these interactions were previously known, such as stronger binding of an oligosaccharyltransferase subunit to wild-type PS1 rather than to the A431E mutant (see Jun 2010 news).

Roughing Up the Eye.
Drosophila eyes (left) degenerate in flies expressing mutant protein ATXN1-Q82 (middle). Knocking down proteins that interact with ATXN1-Q82 hastens destruction, causing purple necrotic spots (right). [Image courtesy of Cell Reports, Hosp et al.]
To test whether interacting proteins coming up in the screen were meaningful, the authors validated a subset of them using a variety of methods, including co-immunoprecipitation. For the 97 putative AD gene interactors, the authors examined GWAS data and found that SNPs near these genes tended to associate with AD, although none reached genome-wide significance. In the case of ataxin-1, the authors expressed the mutant version with an expanded CAG repeat, ATXN1-Q82, in fly eyes, where the gene caused neurodegeneration leading to the rough-eye phenotype. The researchers then knocked down putative interacting proteins in these flies using small hairpin RNAs. In nine of 12 cases, this worsened degeneration of the eye, confirming that these genes contribute to pathology. As a control, the researchers knocked down the same 12 genes in a fly that expressed mutant human tau and had a similar degenerative eye. None of the genes worsened the phenotype, highlighting that these interactors are specific for ATXN1-Q82.
The authors chose one protein for follow-up. The leucine-rich pentatricopeptide repeat motif containing protein (LRPPRC) resides in the mitochondrial matrix, where it binds RNA and regulates translation of mitochondrial genes. More of it bound to APPSwe than wild-type APP. Previous studies reported that APP can insert itself into the mitochondrial membrane, where it supposedly sits with its N-terminal domain sticking down into the matrix (see Keil et al., 2004; Devi and Anandatheerthavarada, 2010). Might LRPPRC bind that N-terminal domain? Supporting this, LRPPRC failed to bind an N-truncated version of APP, the authors found.
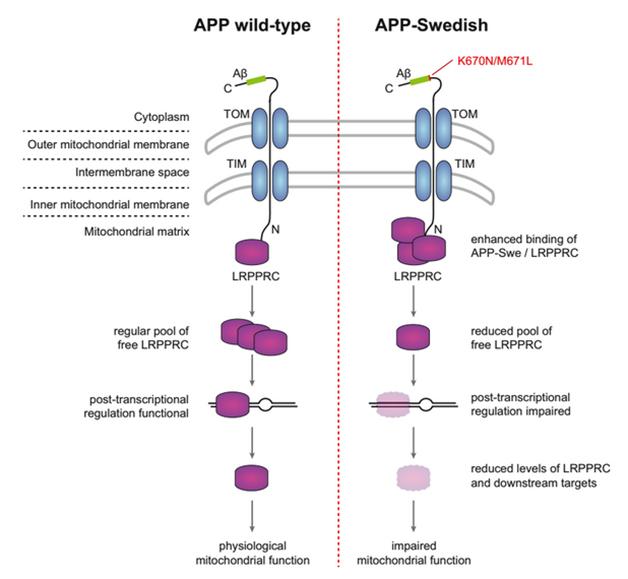
Mutant APP Hampers Mitochondria. In the proposed model, excess APPSwe in the mitochondrial membrane ties up LRPPRC, keeping it from its duties as a regulator of mitochondrial proteins. [Image courtesy of Cell Reports, Hosp et al.]
Curiously, the Swedish mutation occurs closer to the C-terminal end of APP, not the N-terminus. How, then, does it affect binding to LRPPRC? One possibility is that the mutation causes more APP to travel to mitochondria, where it can then bind LRPPRC, the authors suggest. This in turn might keep LRPPRC from promoting protein translation (see image above).
To test this model, the authors transfected cultures with either APP or APPSwe. Cells expressing the mutant protein had much less LRPPRC and lower levels of several mitochondrial proteins regulated by LRPPRC, including cytochrome oxidase, the last enzyme in the electron transport chain. The mitochondria functioned less well in these cells; however, co-expression of LRPPRC largely rescued downstream proteins and mitochondrial activity, supporting the model.
The APPSwe/LRPPRC association points to a role for mitochondrial dysfunction in some forms of familial AD, and is consistent with other work, said Russell Swerdlow at the University of Kansas Medical Center. He noted that Tg2576 mice, which carry APPSwe, develop mitochondrial problems at about 2 months of age, long before amyloid begins to deposit. At that age, genes involved in mitochondrial energy metabolism are upregulated, as though they are compensating for something (see Reddy et al., 2004).
What about sporadic disease? Since LRPPRC interacts with wild-type APP, this pathway might be relevant there, too, the authors speculate. Flint Beal at Cornell University, New York, pointed out that in AD brains, excess APP appears in the mitochondrial membrane, associated with the TOMM40 import channel (see Devi et al., 2006). AD brain tissue often has scant cytochrome oxidase as well. The “mitochondrial cascade hypothesis” of Alzheimer’s suggests that mitochondrial dysfunction acts as a final common pathway in both early and late-onset forms of the disease (see Swerdlow and Khan, 2004).—Madolyn Bowman Rogers
References
Webinar Citations
News Citations
- NIH Funds Prevention Trials and Translational Studies
- Exome–Network Combination Uncovers New Disease Genes
- Death of the Neatnik: Neurons Perish When Trash Clutters Their Space?
Research Models Citations
Paper Citations
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005 Oct 20;437(7062):1173-8. Epub 2005 Sep 28 PubMed.
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O'Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogue K, Colwill K, Gladwish K, Muskat B, Kinach R, Adams SL, Moran MF, Morin GB, Topaloglou T, Figeys D. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. Epub 2007 Mar 13 PubMed.
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011 Jan;39(Database issue):D561-8. Epub 2010 Nov 2 PubMed.
- Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Müller-Spahn F, Haass C, Czech C, Pradier L, Müller WE, Eckert A. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004 Nov 26;279(48):50310-20. PubMed.
- Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer's and Parkinson's diseases. Biochim Biophys Acta. 2010 Jan;1802(1):11-9. PubMed.
- Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004 Jun 15;13(12):1225-40. PubMed.
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006 Aug 30;26(35):9057-68. PubMed.
- Swerdlow RH, Khan SM. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med Hypotheses. 2004;63(1):8-20. PubMed.
Further Reading
News
- Aβ and Mitochondria—When It Reigns, They Pore
- NO Kidding? Mitochondria Fission Protein Linked to Neurodegeneration
- High Cholesterol Leaves Mitochondria Defenseless Against Aβ
- Mitochondrial Break-up: Alzheimer’s Alters Fusion, Fission
- New Triple Transgenic Shows Mitochondrial Damage by Tau, Aβ
- Do Mothers’ Mitochondria Magnify Dementia Risk?
- Honolulu: Tomm40 Reported to Track With Brain Atrophy, Cognition
- A BAD Mitochondrial Dehydrogenase—A Good AD Drug Target?
- Ironing Out Apoptotic Role for New APP-Binding Protein
- So Immature—Aβ Stymies Mitochondrial Protein Processing
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- Studies Suggest Mitochondria Changes Precede Aging, Alzheimer’s
Primary Papers
- Hosp F, Vossfeldt H, Heinig M, Vasiljevic D, Arumughan A, Wyler E, Genetic and Environmental Risk for Alzheimer’s Disease GERAD1 Consortium, Landthaler M, Hubner N, Wanker EE, Lannfelt L, Ingelsson M, Lalowski M, Voigt A, Selbach M. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015 May 19;11(7):1134-46. Epub 2015 May 7 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Harvard Medical School
The report of Hosp and colleagues describes an elegant and novel method for quantitative interaction proteomics with the identification of multiple interactors for neurodegenerative disease associated proteins. A limitation of the approach, as the authors point out, is that it initially requires overexpression of the bait protein by transfection. However, a number of the interactors were subsequently validated by analysis of endogenously expressed proteins. One the most interesting interactions is between APP and LRPPRC, a central regulator of mitochondrial gene expression. This raises the intriguing possibility that mutations and copy number variants of APP that give rise to AD may act in part through mitochondrial dysregulation.
This report provides many potential avenues for future investigation. More broadly, interaction proteomics provides a general and much-needed approach for rapid functional characterization of disease-related genes emerging from GWAS and exome sequencing studies. To validate this approach, follow-up studies using mouse models and other model systems will be needed to confirm the disease relevance of the newly identified interactors.
Make a Comment
To make a comment you must login or register.