Without TREM2, Microglia Run Out of Gas
Quick Links
Loss-of-function variants in the microglial protein TREM2 increase the risk for late-onset Alzheimer’s disease. Now science suggests a reason: Deletion of TREM2 causes a metabolic meltdown for microglia, whereby the cells cannot meet their own energy demands and undergo apoptosis.
In the August 10 issue of Cell, Marco Colonna and colleagues at Washington University in St. Louis show that microglia lacking TREM2 have global changes in their metabolism that result in lowered ATP levels and signs of stress and death. Microglia in these mice were too moribund to ring around amyloid plaques or prevent amyloid toxicity to the surrounding neurons. Correcting this “metabolic derailment,” as the authors call it, with dietary cyclocreatine raised microglial ATP levels. Lo and behold, the cells once again rallied around amyloid plaques, where they protected neurons from toxicity.
“In this extremely exciting paper, the Colonna lab shows that TREM2-deficient dysfunctional microglia can be therapeutically modulated to regain their original defense mechanisms,” wrote Christian Haass and Gernot Kleinberger of the German Center for Neurodegenerative Diseases in Munich.
The Haass lab’s own recent analyses of microglia from TREM2-knockout mice painted a picture of immune cells stuck in neutral with an apparent deficit in glucose uptake. Held back in a quiescent state, the cells were unable to migrate normally, or respond to amyloid (see May 2017 news). Alas, the depiction in this new study is even worse—the microglia are not just sleeping, they are actually quite ill.

Energetic defense.
Amyloid plaques (blue) bloom unbothered by microglia in TREM2-deficient AD mice (left); cyclocreatine treatment (right) reinvigorates woebegone microglia (red) to express activation markers (green) and surround plaques. [Courtesy of Cell, Ulland et al., 2017.]
Microglia, the resident immune cells of the brain, are emerging as key defenders against the damage caused by amyloid plaques. Microglia normally form a protective barrier around amyloid deposits, which prevents plaque spread and shields nearby neurons from toxicity. Pathogenic TREM2 mutations or TREM2 knockout impair this protective function (May 2016 news). However, exactly how loss of TREM2 immobilized microglia was unclear.
To get at that question, first authors Tyler Ulland and Wilbur Song isolated microglia from TREM2 knockout mice crossed with 5XFAD mice, a model that develops early and abundant plaque pathology with neuronal death. Electron micrograph images revealed microglia loaded with multilamellar vesicles resembling autophagosomes. In brain tissue, more than 80 percent of microglia stained positive for the autophagosome marker LC3, compared with 20 percent in either the TREM2 knockout or 5XFAD parental strains. The investigators saw a similar increase in LC3 in postmortem human tissue: Microglia in brains from AD patients carrying the R47H or R62H TREM2 risk allele had three to four times more LC3+ microglia than cases without the risk variants.
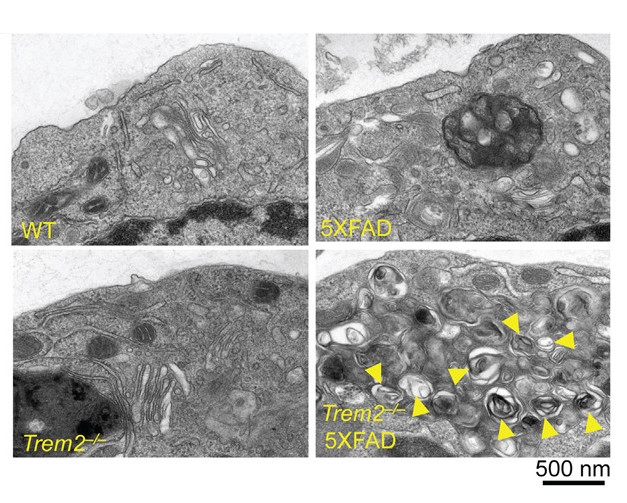
Signs of stress: Autophagosomes (yellow arrows) proliferate in microglia when TREM2 is knocked out in the 5XFAD AD mouse model. [Courtesy of Cell, Ulland et al., 2017.]
Autophagosomes are telltale signs of metabolic stress, where cells cannot keep up with the energy demands of proliferation. Looking more closely at brain microglia ex vivo, the investigators found a significant deficit in activity in the mammalian target of rapamycin (mTOR) pathway, a crucial mediator of cell metabolism and proliferation. In isolated microglia, they found impaired phosphorylation of mTOR pathway intermediates, indicative of deficient mTOR activation and a low energy state in the cells. Compared to microglia from wild-type or 5XFAD mice, the TREM-deficient cells had lower mitochondrial mass, and changes in gene expression tied to aberrant glycolysis and protein synthesis. More of the microglia contained traces of cleaved caspase3, a marker for apoptosis.
Cells without TREM2 responded haplessly to stress in general. In macrophages derived from the bone marrow of TREM2 knockouts, growth factor deprivation increased autophagy and the same mTOR and metabolic failures as seen in the microglia from TREM2-/-5XFAD mice. Metabolomics analysis showed widespread decreases of metabolic intermediates involved in nucleotide synthesis, glycolysis, and energy production, along with more anabolic metabolites.
The results suggest that TREM2 provides trophic support for macrophages and microglia in stressful conditions. “In AD, microglia undergo prolonged stress and activation of proliferation, and that is sustained by mTOR signaling. If there is no TREM2, all the functions of microglia, and ultimately survival, are compromised,” Colonna explained. This suggests that microglia in TREM2 knockout mice are not simply ignoring plaques. “They are being actively driven into a stressed state that is normally compensated by TREM2-depending survival signals,” he told Alzforum
In the short term, autophagy may facilitate clearing of plaque by stimulating microglial phagocytosis, Colonna said. But in the long term, microglia lacking TREM2 die, and that may open the gates to further spreading of plaques.
Can the cells be saved? To try, the researchers turned to cyclocreatine, a compound that raises ATP levels in cells. Treating bone-marrow-derived macrophages in vitro with cyclocreatine raised their metabolic rate, lowered the number of autophagic vesicles, and restored mTOR signaling and cell viability. To treat mice, the investigators added cyclocreatine to the drinking water continuously from 10 weeks of age. At eight months of age, they saw fewer microglia containing autophagosomes, less LC3 staining, and less caspase 3 cleavage in treated mice compared with untreated littermates. Cyclocreatine treatment increased the number of microglia clustered around plaques, and the expression of the microglial activation marker osteopontin.
Importantly, cyclocreatine also improved the cells’ barrier function. In TREM2-/- 5XFAD mice, plaques were diffuse and not surrounded by microglia. After treatment with cyclocreatine, more microglia appeared around plaques, which were denser, resembling those in parental 5XFAD mice. Cyclocreatine treatment did not change the number of plaques or the engulfment of amyloid by microglia, but it did reduce the number of dystrophic neurites around plaques.
The work provides a “proof of principle that if we can rescue the metabolic pathway, somehow we can restore the function of microglia,” Colonna said. “I hope this will inspire pharmacology studies and focus attention on this pathway.”
Cyclocreatine’s relative, creatine, is sold over the counter in the United States and widely used by athletes as a nutritional supplement. Colonna strongly advises against people trying creatine to ward off AD. The activity he sees in mice is no proof of activity in people, and the compound can harm the heart and kidneys with long-term use.—Pat McCaffrey
References
News Citations
- Paper Alert: TREM2 Crucial for Microglial Activation
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
Other Citations
Further Reading
Papers
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016 May 18;90(4):724-39. PubMed.
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016 May 2;213(5):667-75. Epub 2016 Apr 18 PubMed.
Primary Papers
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell. 2017 Aug 10;170(4):649-663.e13. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
ISAR Bioscience
In this extremely exciting paper, the Colonna lab shows that TREM2-deficient, dysfunctional microglia can be therapeutically modulated to regain their original defense mechanisms. Ulland and colleagues show this by restoring energy supply via stimulation of an alternative signaling pathway similar to TREM2. Apparently this also allows the microglia to switch at least partially from a locked homoeostatic state (Mazaheri et al., 2017) to a biologically active defense state.
This even reduces neuritic dystrophies around amyloid plaques, although plaque load itself remains unchanged. The function of TREM2 in amyloid plaque clearance therefore still remains unclear, and may have to be resolved in more physiological animal models with less aggressive amyloid plaque pathology.
Excitingly, TREM2 seems to be located in a central signaling pathway for microglial energy metabolism. The latter is consistent with our recent findings demonstrating reduced FDG-µPET signals in TREM2 p.T66M mice (Figure 1; Kleinberger et al., 2017; see also May 2017 news).
Figure 1. Reduced FDG signal in several brain areas in TREM2 p.T66M knock-in mice at 12 months of age when compared to age- and sex-matched wild-type controls. [Courtesy of Kleinberger et al., 2017.]
All together, these data suggest a major defense function of microglia during disease development. This is also supported by the finding that microglia become activated during healthy aging (Kleinberger et al., 2017); and by the fact that TREM2 is upregulated around amyloid plaques.
This is in line with the finding of increased levels of sTREM2 in CSF of aging people, which are even further enhanced during disease progression (Suárez-Calvet et al., 2016; Suárez-Calvet et al., 2016).
References:
Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trümbach D, Wurst W, Brunner B, Bultmann S, Tahirovic S, Kerschensteiner M, Misgeld T, Butovsky O, Haass C. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017 Jul;18(7):1186-1198. Epub 2017 May 8 PubMed.
Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, Focke C, Deußing M, Suárez-Calvet M, Mazaheri F, Parhizkar S, Pettkus N, Wurst W, Feederle R, Bartenstein P, Mueggler T, Arzberger T, Knuesel I, Rominger A, Haass C. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 2017 Jul 3;36(13):1837-1853. Epub 2017 May 30 PubMed.
Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C, Dominantly Inherited Alzheimer Network. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016 Dec 14;8(369):369ra178. PubMed.
University of Oklahoma Health Sciences Center
This is an exciting paper showing a novel molecular mechanism by which TREM2 alters microglial metabolism. Although we know that TREM2 mutations associated with AD change microglial proliferation and survival, and contribute to plaque build-up and neuron injury, we have limited understanding of the cellular signaling responses in normal or TREM2-deficient microglial to fully explain the increase in AD.
This study shows microglia expressing TREM2 successfully increase mTOR signaling in response to a variety of cellular stresses, thus allowing cells to have metabolic flexibility during times of stress. In the absence of TREM2, the microglia fail to upregulate mTOR and instead have activation of dysregulated autophagy, leading to decreased ability to have glycolytic capacity.
Interestingly, the data supports a tonic TREM2 signal needed for basal mTOR activation, and during cellular stress there appears to be an increased requirement for TREM2-induced mTOR to allow microglial proliferation and survival. Thus, the stressed microglial cells become increasingly dysfunctional because of the loss of metabolic flexibility.
Dysregulated mTOR is already known to play a role in AD, Down’s syndrome, Huntington’s chorea, and other neurological disorders, but has not previously been linked to TREM2.
Interestingly, the authors show that bypassing TREM2 by providing a direct energy analog, cyclocreatine, can rescue the abnormal TREM2-deficient microglial dysfunction in vitro and limit neuronal dystrophy in response to Aβ plaques in an AD mouse model. This is exciting data, but as the authors correctly point out, additional studies are needed before cyclocreatine is used in patients with AD as patients may experience a variety of health problems, including changes in blood glucose, muscle cramping, and weight gain. Additionally, cyclocreatine may interact with commonly used medications. However, these results will may pave the way for new treatment options to prevent or treat AD.
Yale University
This is an interesting paper that uncovers an unexpected molecular mechanism linking TREM2 deficiency to microglia metabolic state through the mTOR pathway. It is interesting that the metabolic-deficient phenotype is mostly seen in microglia that are clustering around amyloid plaques but less so in those not engaged with plaques. This suggests that the state of activation of microglia (associated with upregulation of TREM2) and their polarization toward plaques may be energetically demanding and renders these cells more susceptible to homeostatic disruptions.
We have recently reported that the polarization of microglial processes toward plaques, leading to their encapsulation, is a very important function independent of phagocytosis. Plaque encapsulation modifies the degree of plaque compaction (and thus their toxicity), and by insulating plaques from adjacent neuronal processes acts as a neuroprotective barrier (Condello et al., 2015; Yuan et al., 2016; Wang et. al. 2016).
We found that TREM2 deficiency in mice or the R47H variant in humans lead to reduced plaque encapsulation, which changes amyloid fiber conformation to a less compact and potentially more toxic one. Indeed, we found that this was associated with a marked increase in axonal dystrophy and tau hyperphosphorylation around plaques that have deficient microglia encapsulation, suggesting that the loss of microglia’s protective function led to increased neuronal process injury.
Consistent with this view, Ulland et al. found that the reduced clustering of microglia around plaques is associated with increased axonal dystrophy and that the administration of cyclocreatine to ameliorate the metabolic deficit in microglia improves clustering and reduces axonal dystrophy.
Overall, this study provides a novel mechanism explaining the failure of microglia plaque engagement in TREM2 mutants. A similar metabolic deficit could occur in aging, where we have shown that microglia plaque encapsulation becomes deficient and axonal dystrophy is increased (Condello et al., 2015). Thus, this paper could have implications beyond TREM2 mutants, for the highly prevalent late-onset AD.
References:
Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015 Jan 29;6:6176. PubMed.
Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016 May 18;90(4):724-39. PubMed.
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016 May 2;213(5):667-75. Epub 2016 Apr 18 PubMed.
Make a Comment
To make a comment you must login or register.