Semagacestat Failure Analysis: Should γ-Secretase Remain a Target?
Quick Links
When a large trial fails, there is usually hand wringing, doom saying, and not a little bit of Monday-morning quarterbacking. But beyond the din of disappointment, are we taking enough time to truly learn what went wrong and what could be done better next time? And are we learning publicly, for the benefit of all? At the NIH Alzheimer’s Disease Research Summit 2015—and in meetings and conversations before and since—leading scientists from both academia and industry have called for deeper efforts to dissect all aspects of a terminated drug program and use the insight to push the field forward.
Bart De Strooper of KU Leuven, Belgium, made such an attempt. He struck a nerve, and a probing discussion ensued on Alzforum. In a Leading Edge Essay in the November 6 Cell, De Strooper claimed that the field at large had insufficiently dissected why the semagacestat IDENTITY Phase 3 trial failed. He charged that the field jumped to a simplistic conclusion in abandoning γ-secretase as a target. Specifically, De Strooper argued that the biphasic pharmacology of semagacestat seen in Phase 2 predicted that the dosing regimen chosen for Phase 3 might be counterproductive. The drug caused peaks of complete inhibition alternating with full enzyme activity, rather than a more desirable partial but chronic inhibition, and the once-daily dosing driven by safety concerns worsened this oscillating pattern.
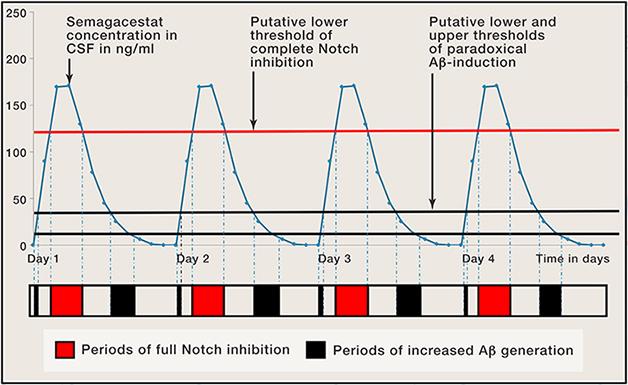
Did semagacestat concentration, Notch inhibition, and Aβ generation fluctuate in clinical trial participants? [Image courtesy of Cell, 2014.]
More broadly, however, De Strooper believes that this and other data could fill key knowledge gaps toward more specific attempts at inhibiting γ-secretase in the future. He urged that the enzyme complex be kept as a therapeutic target. At the very least, he said, the semagacestat results should be followed up with new basic research. To Alzforum, De Strooper wrote: “We have only two classical drug targets in Alzheimer’s, BACE1 and γ-secretase. We should be careful with them.”
In response, 12 scientists developed a thoughtful conversation (see below) about semagacestat and γ-secretase. These contributors run the gamut from academia to biopharma, from basic Notch to clinical trials research. All have worked on specific aspects of γ-secretase or semagacestat. To enable a broader conversation in the field of Alzheimer’s research, Cell Press has generously agreed to open access to De Strooper’s essay for several weeks. Pour yourself a large mug of coffee, read the essay, and take in the considered view of leaders in the field. What do you think? Does this experience with an early, broad-spectrum inhibitor mean this target is too complex? What should be done next to prepare the ground for a better approach? Weigh in with your expertise. We welcome comments about new angles, both in support or in cordial dispute.—Gabrielle Strobel
References
Therapeutics Citations
External Citations
Further Reading
No Available Further Reading
Primary Papers
- De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell. 2014 Nov 6;159(4):721-6. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University School of Medicine
I agree with Bart de Strooper that careful analysis of the semagacestat trial would greatly improve understanding of γ-secretase, a key target in Alzheimer’s disease. Key scientific questions include:
In particular, understanding these scientific questions will enable the field to potentially advance drugs that target γ-secretase in a more informed approach. Related approaches such as γ-secretase modulators, APP-selective GSIs, and GSIs with alternative PK/PD profiles are highly promising and should be fully pursued as potential drugs for AD.
We should remember the medical history lessons from the development of statins. These drugs were almost lost due to early concerns of toxicity (see P. Roy Vagelos, 2004, Medicine, Science and Merck, Cambridge University Press, and Akira Endo, 2010).
The essence is summarized in this citation: “But Merck discontinued statin development upon learning that Sankyo abandoned its statin program after discovering what seemed to be cancerous changes in experimental animals fed large statin doses. Despite a well-established association between high LDL cholesterol levels and cardiovascular complications, researchers worried that reducing blood cholesterol would cause side effects, because cholesterol is an essential component of body cells.
"Statin development stalled for three years until Edward Scolnick assumed a research leadership role at Merck. Scolnick devoted a large fraction of Merck's research budget to overcoming concerns about statin toxicity, and the results convinced the Food and Drug Administration that the findings that killed Sankyo's program were not really cancers and that proceeding with human trials of stain therapy was reasonable” (Boston Globe 2008).
References:
Endo A. A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86(5):484-93. PubMed.
View all comments by Randall BatemanGoizueta Institute @ Emory Brain Health
Bart De Strooper’s essay in Cell is a thoughtful evaluation of a “failed” γ-secretase inhibitor (GSI) trial with semagacestat. I agree that the clinical trials with this compound could have been conducted in ways that would provide more guidance for future studies, but would say that the same is true of many of the previous failed Alzheimer’s disease therapeutic trials. Too often data that could have been used to guide future studies was not collected, and thus negative trials fail to fully inform the next generation of therapeutic development. From my perspective, one of the most problematic studies in this regard is the Phase 2 study of the active Aβ1-42 vaccine with QS21 adjuvant known as AN1792. This trial was halted due to both a low percentage of responders in terms of antibody titers and the unexpected development of meningoencephalitis. During the trial, critical information on T-cell responses was not obtained. Though some ancillary data would support the inference that an auto-reactive T-cell response against the T-cell epitope in Aβ could be linked to the meningoencephalitis, this has never been proven. Nevertheless, this inference became the underlying premise for the vast majority of subsequent active vaccine development. Given the ongoing interest in Aβ vaccines, it would have been preferable to have more data to guide future development.
Given the incredible resources needed to develop and evaluate any new therapy for AD, it is clear in retrospect that the field could have done better. However, clinical trials are constrained in scope by many practical issues. Most of us are not privy to the internal company discussions that drove the development plan, and I personally think we should be cautious about retrospectively saying, “The trial should have been done differently.” It is, however, useful to say, as Dr. De Strooper does in his essay, “It would have been great if the trial had collected this type of data …” This kind of dialogue can help the field conduct a better trial in the future.
Therefore it is extremely encouraging to see the novel public-private partnerships behind the three recently launched prevention studies (API, DIAN, A4, see Dec 2014 news). Not only are these much better tests of the amyloid hypothesis (Golde et al., 2011), but there is tremendous input from academic and industry partners on trial design and data to be collected. Further, from my understanding, there will be an almost unprecedented degree of open access to various aspects of the data obtained from these trials. This prospective plan for data sharing is truly a welcome change. We all hope that these trials yield positive news, but irrespective of clinical outcome they no doubt will provide incredibly valuable data for future “prevention” trials.
Back to Dr. de Strooper’s essay. He raises many points that collectively make a reasonable and rational argument that maybe it is too early to throw in the towel on future development of GSIs for use in Alzheimer’s disease. At a theoretical level, I wholeheartedly endorse this position. It is certainly far too soon to abandon academic efforts at a deeper understanding of γ-secretase biology that could one day lead to the identification of optimized drugs targeting this enzyme, even if personally I remain biased toward a modulator approach for AD as being preferable to a GSI. From my perspective, the focus of this ongoing effort needs to be expanded to the broader biology of γ-secretase and use of GSIs in other disorders.
From a practical viewpoint, with numerous promising BACE inhibitors now in the clinic, I think it is unlikely that we will see a lot of further development of GSIs for AD by the private sector. BACE inhibitors should test the hypothesis of whether inhibiting Aβ production can have therapeutic efficacy. As opposed to current GSIs, at least some BACE inhibitors appear to have greater engagement in the CNS and so far have been well-tolerated.
Outside of the CNS, therapeutic inhibition of γ-secretase has been most often associated with reduced Notch 1 signaling; GSIs are often thought of in these settings as “Notch 1 inhibitors.” Indeed, γ-secretase has now been proposed as a target in various cancers, immunologic disorders including graft versus host disease, vasculitis, macular degeneration, diabetic nephropathy, ischemic reperfusion injury in the kidney, ischemic stroke, traumatic brain injury, hearing loss, and fibrosis (Golde et al., 2013). A main focus of the repurposing of GSIs has been in cancer, with more than 40 human trials underway, terminated, or recruiting. Notably, although some of the expected side effect profile has been observed in these trials (e.g., diarrhea) , there has been good evidence in some of these studies for Notch1 inhibition. Further, altered dosing regimens with GSI treatment for a few days “on” followed by a few days “off” has been key to avoiding toxicities (Richter et al., 2014; Lee et al., 2014; Sahebjam et al., 2013; Diaz-Padilla et al., 2013; Strosberg et al., 2012; Krop et al., 2012; Kolb et al., 2012; Wei et al., 2010; Schott et al., 2013).
One important question for these ongoing studies is whether all current GSIs are biologically equivalent. Though many GSIs currently in cancer trials are considered “pan-GSI inhibitors,” this labeling may be a misnomer. GSI inhibitory activity is often only established for Aβ and Notch 1. Net action of GSIs may be influenced by multiple factors within a target cell. These factors not only include the variable subunit composition of the γ-secretase complexes, but also a) expression and location of substrate and enzyme in the target cell, b) sheddase expression, and c) activation of the sheddase. Clearly, given the current investment in repurposing GSIs, additional studies directly comparing biological actions of various GSIs used in clinical trials in various model systems are warranted. Indeed, the repurposing effort is exciting. It has a solid rationale and, for certain cancers where the data is published, appears to be based on solid preclinical data (reviewed in Groth and Fortini, 2012).
Clearly γ-secretase and other biomedically important intramembrane cleaving proteases (e.g., SPPs) are fascinating proteins that play significant roles in numerous aspects of human physiology and pathophysiology. For those of us who have made long-term investments in the study of these enzymes, it can be disheartening to see our ongoing work deprioritized because a pan-GSI might not work for Alzheimer’s. Hopefully, through thoughtful discourse such as initiated here by Dr. De Strooper, we can make the case that the fat lady has not yet sung the blues on γ-secretase.
References:
Golde TE, Schneider LS, Koo EH. Anti-aβ therapeutics in Alzheimer's disease: the need for a paradigm shift. Neuron. 2011 Jan 27;69(2):203-13. PubMed.
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013 Jun 17; PubMed.
Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M, Chen Z, Cohen B, Zhang WJ, Wang L, Ivy SP, Moore MJ, Oza AM, Siu LL, McWhirter E. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs. 2014 Apr;32(2):243-9. Epub 2013 May 5 PubMed.
Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, Othus M, Ribas A, Sondak VK, Gajewski TF, Margolin KA. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015 Feb 1;121(3):432-40. Epub 2014 Sep 23 PubMed.
Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T, Kamel-Reid S, Chen H, Ivy SP, Razak AR, Oza AM, Chen EX, Hirte HW, McGarrity A, Wang L, Siu LL, Hotte SJ. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer. 2013 Aug 20;109(4):943-9. Epub 2013 Jul 18 PubMed.
Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ, Razak AA, Chen EX, Brana I, Wizemann M, Wang L, Siu LL, Bedard PL. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2013 Oct;31(5):1182-91. Epub 2013 Jul 17 PubMed.
Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H, Sullivan D. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012 May;48(7):997-1003. Epub 2012 Mar 23 PubMed.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012 Jul 1;30(19):2307-13. Epub 2012 Apr 30 PubMed.
Kolb EA, Gorlick R, Keir ST, Maris JM, Lock R, Carol H, Kurmasheva RT, Reynolds CP, Kang MH, Wu J, Houghton PJ, Smith MA. Initial testing (stage 1) by the pediatric preclinical testing program of RO4929097, a γ-secretase inhibitor targeting notch signaling. Pediatr Blood Cancer. 2012 May;58(5):815-8. Epub 2011 Aug 16 PubMed.
Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, Randolph S, Smeal T. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010 Jun;9(6):1618-28. Epub 2010 Jun 8 PubMed.
Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013 Mar 15;19(6):1512-24. Epub 2013 Jan 22 PubMed.
Groth C, Fortini ME. Therapeutic approaches to modulating Notch signaling: current challenges and future prospects. Semin Cell Dev Biol. 2012 Jun;23(4):465-72. Epub 2012 Jan 30 PubMed.
View all comments by Todd E. GoldeUniversity of Goteborg, Sahlgrenska University Hospital
University of Gothenburg
This is an interesting article, and we agree with its scientific argument. Bart de Strooper is right about the short half-life of semagacestat, and important aspects such as its rebound effect on CSF Aβ levels. Whether this explains the cognitive worsening seen in the trial is just a hypothesis at this point. There could be several other explanations, maybe including inhibition of Notch and also other γ-secretase substrates.
γ-secretase should not be abandoned as a pharmacological target. However, there is more data in favor of activating γ-secretase than inhibiting it. Bart's team themselves have shown in beautiful studies that most PSEN1 mutations lead to a less effective γ-secretase (Chávez-Gutiérrez et al., 2012). It manages to produce Aβ1-42 and 1-40 from APP, but less so the shorter Aβ1-37/38/39 isoforms that may be protective and inhibit Aβ1-42 oligomerization/fibrillization. We have shown similar results for the PSEN1 A431E mutation (Portelius et al., 2010). A GSA or GSM that boosts cleavages at Gly37, Gly38 and Val39 should be a good candidate to reduce the risk of cerebral β-amyloidosis. We clearly have more to learn about the action of GSIs.
References:
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Portelius E, Andreasson U, Ringman JM, Buerger K, Daborg J, Buchhave P, Hansson O, Harmsen A, Gustavsson MK, Hanse E, Galasko D, Hampel H, Blennow K, Zetterberg H. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer's disease. Mol Neurodegener. 2010;5:2. PubMed.
View all comments by Erik PorteliusAsceneuron SA
Bart De Strooper’s essay provides a well-written analysis and refreshing views on the publicly available data on the γ-secretase inhibitor semagacestat. I could not agree more with him that γ-secretase is a fascinating enzyme and academic research needs to continue, as it is poised to lead to further exciting discoveries. I am less optimistic, however, about a potential revival of this protease as a target for Alzheimer’s drug discovery. The case presented for semagacestat highlights the importance of peak and trough drug levels in addition to the overall drug exposure. This makes particular sense when considering oscillating systems as highlighted in the manuscript. It is noteworthy that semagacestat is only one out of many γ-secretase inhibitors that have failed in the development for Alzheimer’s disease. Given the structural diversity of γ-secretase inhibitors (Olson and Albright, 2008), one can presume that each of them had its own characteristic pharmacokinetic/pharmacodynamic profile and most γ-secretase inhibitors have never progressed beyond Phase 1 safety, tolerability and pharmacodynamic studies.
The main consideration had to be whether there were means to achieve the desired reduction in Aβ peptide production without causing Notch-related side effects, in many instances manifested by gastrointestinal toxicities. In this respect, avagacestat is another example of a late-stage clinical failure despite tremendous efforts to identify a γ-secretase inhibitor that was reported to have an intrinsic preference for the inhibition of the processing of APP over the Notch receptor (Mayer et al., 2008). This left the field with Notch-sparing γ-secretase modulators, which at the time of their discovery appeared to provide the solution to this key problem (Weggen et al., 2001).
A large number of drug-like γ-secretase modulators have been reported in the past years, but only very few of them are still being pursued in the clinic. Usually this would imply that this class of compound also encountered some sort of difficulty in late-stage preclinical or early clinical development. The high lipophilicity of these molecules has already been identified as one of the issues (Gijsen and Mercken, 2012).
In my view, the way Alzheimer’s drugs are still being developed imposes the greatest hurdle to future γ-secretase drug development efforts, including those addressing the main suggestion from Bart De Strooper’s essay. Generally, it requires an investment of hundreds of millions of U.S. dollars to enter the seminal proof-of-concept stage that tends to feature a large Phase 3 clinical trial. Thus, without the confidence that a reduction of Aβ peptide production could lead to a meaningful therapeutic benefit, one can assume most companies will shy away from such an endeavor for a target that was previously shown to be troublesome. Nevertheless, a positive outcome in one of the current BACE1 inhibitors trials could potentially reinvigorate the interest in γ-secretase-targeting modalities. On the other hand, in today’s world of drug development, with ever-growing pressure of pharmacoeconomics on decision-making, this would require a careful analysis of the commercial potential of such a drug compared to BACE1 inhibitor.
References:
Olson RE, Albright CF. Recent progress in the medicinal chemistry of gamma-secretase inhibitors. Curr Top Med Chem. 2008;8(1):17-33. PubMed.
Mayer SC, Kreft AF, Harrison B, Abou-Gharbia M, Antane M, Aschmies S, Atchison K, Chlenov M, Cole DC, Comery T, Diamantidis G, Ellingboe J, Fan K, Galante R, Gonzales C, Ho DM, Hoke ME, Hu Y, Huryn D, Jain U, Jin M, Kremer K, Kubrak D, Lin M, Lu P, Magolda R, Martone R, Moore W, Oganesian A, Pangalos MN, Porte A, Reinhart P, Resnick L, Riddell DR, Sonnenberg-Reines J, Stock JR, Sun SC, Wagner E, Wang T, Woller K, Xu Z, Zaleska MM, Zeldis J, Zhang M, Zhou H, Jacobsen JS. Discovery of begacestat, a Notch-1-sparing gamma-secretase inhibitor for the treatment of Alzheimer's disease. J Med Chem. 2008 Dec 11;51(23):7348-51. PubMed.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001 Nov 8;414(6860):212-6. PubMed.
Gijsen HJ, Mercken M. γ-Secretase Modulators: Can We Combine Potency with Safety?. Int J Alzheimers Dis. 2012;2012:295207. PubMed.
View all comments by Dirk BeherUSC Alzheimer’s Therapeutic Research Institute
This excellent article by Bart de Strooper discusses the implications of the negative Phase 3 trial of Semagacestat in mild to moderate AD that led to discontinuation of the development of this compound. He points out that the once-daily regimen studied provided peaks of enzyme inhibition alternating with periods of no inhibition. This dosing may have reduced therapeutic effects, while causing significant toxicity possibly related to effects on Notch signaling. De Strooper bemoans the impact of the Semagacestat failure on clinical and basic research on γ-secretase.
This is an important message: a randomized controlled clinical trial is the best possible test of therapeutic effects, but each trial tests a specific regimen of a specific drug, in a particular population, using specific measures. There is substantial risk in generalizing the results to draw broad conclusions on the therapeutic strategy.
On the other hand, I cannot agree with de Strooper’s view that the trial was launched prematurely. The enormity of the burden of this disease justifies launching trials (even trials as large, long, and costly as the Semagacestat trial; indeed, shorter Phase 2-type studies of candidate disease-slowing agents cannot be very informative in this disease) with an imperfect understanding of the target and the drug’s pharmacodynamics. The regimen selected for the trial reduced Aβ generation as indicated by SILK study; a twice-daily regimen was not feasible because of toxicity. We must make the best decisions based on available information, and conduct our studies of efficacy and safety. We should be investing much, much more in all levels of AD research, from basic science to therapeutic trials, while assuring that we share data and collaborate as fully as feasible to accelerate our progress.
Discontinuation of Semagacestat development was a reasonable decision based on the trial results. But this does not indicate that the amyloid hypothesis is incorrect, nor does it lessen the need for basic and clinical investigation of γ-secretase, which remains among the most plausible therapeutic targets.
View all comments by Paul AisenCo-Director, Brigham and Women's Hospital's Ann Romney Center for Neurologic Diseases
Harvard Medical School and Brigham and Women's Hospital
With the failure of the semagacestat trials, it is easy to quickly dismiss γ-secretase as a viable drug target and move forward to alternative targets for the treatment of Alzheimer’s disease. However, in this letter to Cell, Bart De Strooper provides a detailed analysis of the data resulting from these trials. He makes an excellent case for why we should not abandon γ-secretase just yet, but instead, have renewed focus on better understanding the basic biology of this complex and fascinating protease. We agree with his analysis and overall conclusions and would like to emphasize a few key points regarding the trial data:
1. The rationale for targeting γ-secretase is to decrease the generation of Aβ and reduce the Aβ burden mainly in the brain, where it is deposited as amyloid plaques and also accumulates in synaptotoxic soluble oligomers that are in a complex equilibrium with the plaques. From the Phase 1-3 trials, CSF measures of Aβ were never shown to be significantly reduced, suggesting that semagacestat did not have the desired impact on the steady-state levels of Aβ in the CNS (Siemers et al., 2005; Siemers et al., 2007; Siemers et al., 2006; Doody et al., 2013; Fleisher et al., 2008). Bateman and colleagues (Bateman et al., 2009) were able to show an acute reduction in newly generated Aβ by a single dose of semagacestat, however, its effect on the overall steady-state levels of Aβ is more difficult to conclude.
2. Another important emphasis De Strooper makes is that due to toxicity concerns from a compound with a very low therapeutic index, patients were dosed only once per day. Given the short half-life of the drug (~2.2 to 2.6 hours), patients would have daily fluctuations between high drug concentrations and little or no drug in the plasma (and CSF). In accord, plasma Aβ levels, while initially reduced upon drug administration, quickly returned to baseline and later increased to levels above baseline (Siemers et al., 2005; Siemers et al., 2007; Doody et al., 2013), likely resulting in no overall reduction in Aβ load over time in plasma. Thus, we cannot conclude that semagacestat actually lowered Aβ in the trial.
3. Semagacestat appeared to be more effective at reducing Aβ40 than Aβ42, resulting in an increase in the ratio of Aβ42/40—roughly similar to the effect of pathogenic FAD-causing mutations found in presenilin (Doody et al., 2013).
While the results of the trial were disappointing, these data indicate that the amyloid hypothesis was not rigorously tested. Moving forward, many have begun to look beyond γ-secretase to β-secretase as an alternative method to lower Aβ. However, we believe that β-secretase inhibitors administered chronically to humans will likely develop some adverse effects analogous to their γ-counterpart. β-secretase has been shown to be an active sheddase in mouse primary neurons. It contributes about 19 percent of identified shed proteins (Kuhn et al., 2012), including Neuregulin, which has important functions in myelination (Fleck et al., 2013). Furthermore, chronic inhibition of BACE activity by genetic ablation (Willem et al., 2006) or with prolonged inhibitor treatment (Filser et al., 2014) has resulted in adverse effects in mice. In addition, peptidomimetic inhibitors of BACE are prone to P-gp-mediated efflux and are therefore less BBB-penetrant; however, the latest generation of BACE inhibitors has apparently overcome this issue.
There are still quite a few benefits that targeting γ-secretase could provide over β-secretase. First, γ-secretase can be modulated, as opposed to inhibited, to reduce the relative levels of Aβ42. By shifting γ-secretase processing to favor shorter Aβ isoforms, the generation of soluble intracellular domains of many important substrates such as Notch will not be affected, and this should lessen side effects in humans. Alternatively, selective γ-inhibitors that specifically target APP as opposed to other γ-substrates can be developed; currently, some γ-inhibitors are relatively Notch-sparing. In addition, since γ-secretase is an intramembrane protease, inhibitors and modulators that target this enzyme tend to be cell-penetrant and thus may allow for better BBB permeability.
However, before we can further develop novel approaches to target γ-secretase safely and effectively, more needs to be learned about the basic physiology of the γ-secretase complex and substrate processing.
First, inhibitors of the presenilin/γ-secretase complex need to be better characterized and their mechanism of action better understood. Semagacestat, for example, appears to have a dual nature as both an activator at low concentrations and an inhibitor at high concentrations, suggesting that it might not be a traditional inhibitor per se. Second, do semagacestat (and other γ-inhibitors) inhibit all the various γ-secretase complexes that are composed of variants of presenilin and APH-1 equally, or might there be a preference for certain complexes? In this regard, it is important to determine which complexes (or all) contribute most to Aβ42 generation (perhaps APH-1B containing complexes; see Serneels et al., 2009) and which may contribute most to normal Notch signaling. Identifying whether there is specificity toward certain substrates will allow for more selective inhibition and targeting of specific γ-secretase complexes. Third, it has been shown that FAD-causing PS mutations, which can change the conformation of the presenilin/γ-complex (Berezovska et al., 2005), are more resistant to γ-inhibitors (Xia et al., 2000). If this conformational change also occurs in some sporadic AD subjects to generate pathological γ-complexes that increase the Aβ42/40 ratio, then standard inhibitors may not target these “pathological” complexes as efficiently, but instead target γ-complexes generating normal ratios of Aβ42/40. This may explain why in the Lilly trial, semagacestat was more effective in reducing the levels of Aβ40 than those of Aβ42.
A fourth area of study is understanding the complexity and breadth of γ-secretase substrates and their regulation. Currently, there are well over 70 substrates and likely more to be discovered. While much effort has been focused on the processing of APP and Notch, the functions of other substrates and the physiological importance of their γ-processing will be critical for understanding γ-secretase function and the consequence of its inhibition.
Fifth, how γ-secretase is regulated is another understudied subject. From examining the semagacestat trials (and other studies using various γ-inhibitors), there can be a compensatory increase in the levels of Aβ over baseline upon clearance of the inhibitor. Whether this is due to an active signaling process within the cell that regulates γ-activity or just the clearance of the accumulated β-CTFs remains to be seen. By understanding this potential feedback mechanism better, we can develop drugs or treatment paradigms that have sustained reduction in Aβ generation.
All γ-secretase cleavages (to date) are preceded by a shedding event by either α- or β-secretase. How these various cleavages are coordinated or regulated is another important topic to explore. We have observed in our lab that inhibition of γ-secretase by multiple inhibitors, including semagacestat, can significantly upregulate the activity of α-secretase in living cells, as assayed by an increase in APPsα secretion into the media. This occurred at the expense of β-secretase activity, as APPsβ secretion was decreased. The increase in α-activity that we have observed is consistent with the finding from Portelius and colleagues (Portelius et al., 2010; Portelius et al., 2012) that treatment with semagacestat in humans resulted in an increase in an Aβ15/16 peptide, a likely product of α-secretase processing of C99. These data suggest that the cleavages by γ-secretase and its sheddases may be more interconnected than thought and may cross-regulate each other.
Finally, the AD field needs to be circumspect and patient as new results from disease-modifying clinical trials emerge. It is crucial that we “get it right,” that is, we understand the biochemistry of our targets and their inhibitors in substantial detail and not rush to conclusions about a therapeutic approach before a deep analysis of the human data has occurred.
References:
Siemers E, Skinner M, Dean RA, Gonzales C, Satterwhite J, Farlow M, Ness D, May PC. Safety, tolerability, and changes in amyloid beta concentrations after administration of a gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005 May-Jun;28(3):126-32. PubMed.
Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007 Nov-Dec;30(6):317-25. PubMed.
Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, Satterwhite J, Gonzales C, Dean RA, May PC. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006 Feb 28;66(4):602-4. PubMed.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, , Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341-50. PubMed.
Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thal LJ. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008 Aug;65(8):1031-8. PubMed.
Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009 Jul;66(1):48-54. PubMed.
Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, Haass C, Rossner S, Bräse S, Lichtenthaler SF. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012 Jul 18;31(14):3157-68. PubMed.
Fleck D, van Bebber F, Colombo A, Galante C, Schwenk BM, Rabe L, Hampel H, Novak B, Kremmer E, Tahirovic S, Edbauer D, Lichtenthaler SF, Schmid B, Willem M, Haass C. Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J Neurosci. 2013 May 1;33(18):7856-69. PubMed.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, Destrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006 Oct 27;314(5799):664-6. PubMed.
Filser S, Ovsepian SV, Masana M, Blazquez-Llorca L, Brandt Elvang A, Volbracht C, Müller MB, Jung CK, Herms J. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry. 2015 Apr 15;77(8):729-39. Epub 2014 Oct 29 PubMed.
Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, Esselmann H, Paul S, Schäfer MK, Berezovska O, Hyman BT, Sprangers B, Sciot R, Moons L, Jucker M, Yang Z, May PC, Karran E, Wiltfang J, D'Hooge R, De Strooper B. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009 May 1;324(5927):639-42. Epub 2009 Mar 19 PubMed.
Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, Hyman BT. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005 Mar 16;25(11):3009-17. PubMed.
Xia W, Ostaszewski BL, Kimberly WT, Rahmati T, Moore CL, Wolfe MS, Selkoe DJ. FAD mutations in presenilin-1 or amyloid precursor protein decrease the efficacy of a gamma-secretase inhibitor: evidence for direct involvement of PS1 in the gamma-secretase cleavage complex. Neurobiol Dis. 2000 Dec;7(6 Pt B):673-81. PubMed.
Portelius E, Dean RA, Gustavsson MK, Andreasson U, Zetterberg H, Siemers E, Blennow K. A novel Abeta isoform pattern in CSF reflects gamma-secretase inhibition in Alzheimer disease. Alzheimers Res Ther. 2010;2(2):7. PubMed.
Portelius E, Zetterberg H, Dean RA, Marcil A, Bourgeois P, Nutu M, Andreasson U, Siemers E, Mawuenyega KG, Sigurdson WC, May PC, Paul SM, Holtzman DM, Blennow K, Bateman RJ. Amyloid-β1-15/16 as a Marker for γ-Secretase Inhibition in Alzheimer's Disease. J Alzheimers Dis. 2012 Apr 18; PubMed.
View all comments by Allen ChenUniversity of Wisconsin, Madison
My comments are based on 20-plus years of experience at the bench generating and examining raw data from experiments on Notch and functionally related genes in Drosophila (see Wesley publications on Notch). I started out asking how Notch in Drosophila embryos can suppress neurogenesis at one stage, i.e., lateral inhibition, but promote neurogenesis at the subsequent stage, i.e., differentiation of neuronal precursor cells. The search for answers has led to novel aspects of Notch functions that are relevant to Bart De Strooper’s discussion of the effect of γ-secretase inhibition on Notch. My commentary uses only published or submitted data.
An unbiased examination of the extensive Notch-related literature from about 1960 in Drosophila and from about 1995 in mammals leads to two conclusions. One, many but not all Notch functions are mediated by the Notch intracellular domain (NICD), which is different from the full-length Notch protein produced from the Notch gene (NFull). Two, the majority of research papers and reviews in the field focus on canonical Notch signaling based on NICD activities, as it is the best-understood aspect of Notch functions and the easier one to manipulate experimentally. Unfortunately, our ease is no help when confronted with the full range of in vivo Notch functions, as is the case in a clinical trial. I believe the trajectory of the semagacestat trial would have been different if the Notch field was less biased toward pursuing data that make sense in light of canonical NICD signaling, while ignoring data that does not make sense from this signaling perspective.
What is the range of known Notch activities? Data from my lab and others indicate that Notch activities are bi-phasic. Immediately after ligand binding, a non-canonical PKC-dependent Notch signaling activity right at the cell surface upregulates one set of genes that includes actin regulators (ovo-shavenbaby, β-tubulin), IκB (Cactus), and CREB. Subsequently, upon significant accumulation of NICD in the cell, canonical Notch signaling in the nucleus upregulates a different set of genes that includes the HES family of transcription factors. In addition to being activated in a linked sequence, these two Notch activities are negatively correlated and manifest ultradian oscillation, i.e., they recur within one day. Furthermore, NFull appears to be proteolytically processed to generate smaller fragments that act as natural dominant-negative molecules to downregulate Notch activities in a cell-type-specific manner or perform other activities, such as axon path-finding. Finally, there is evidence suggesting that one or the other Notch activity or dominant-negative molecule predominates in specific regions, tissues, and cell types at all developmental stages.
With regard to mammals, I have examined Notch protein expression and activities (Notch 1, 2, and 3) in cultured cell systems from human and rodent tissues. These data are limited and unpublished. However, Drosophila Notch-like features are decipherable, suggesting that mammalian Notch receptors might also have diverse activities. Indeed, many papers have reported non-canonical Notch activity in mammalian systems and processes.
Regarding De Strooper’s essay, perhaps the most important implication of my data is that Notch, Delta, presenilin (γ-secretase), Kuzbanian (ADAM10), Suppressor of Hairless (RBP-j), PKC, Armadillo (β-catenin), and Dishevelled cannot be studied in isolation from one another. This conclusion is fascinating scientifically, challenging experimentally, and frustrating in terms of securing funding. But clearly, each of these players responds to alteration in others. Of particular significance, treatment of Notch-expressing Drosophila cells with a γ-secretase inhibitor or increased NICD expression in flies reduces NFull produced from the endogenous Notch gene in the background. Consider these responses in light of the fact that NFull is required not only for NICD-dependent signaling but also for the PKC-dependent signaling. The latter signaling promotes CREB expression, enhances memory formation, and IκB Cactus expression, which suppresses immunity. Even a brief pulse of NFull or NICD expression from a transgene perturbs the ultradian oscillation of endogenous Notch gene activity in the background in a dose-dependent manner (as measured by CREB level). Once the pulse of transgenic NFull or NICD expression has dissipated, the endogenous CREB level overcompensates; this is typical of oscillatory systems. Intriguingly, even transgenic NFull and NICD show evidence of ultradian oscillation following a pulse of induction. We think that Notch activities and the natural dominant-negative mechanisms regulating them intrinsically oscillate in vivo.
Based on the above, the semagacestat trial could have multiple connections to Notch, including canonical or non-canonical signaling, disruption of their regulation, and perturbation of Wnt, PKC, and NFκB pathways. From this perspective, staring at Figure 1 in the essay, which depicts possible semagacestat cycling in patients, makes me wonder about the rich underlying biology that could be at play here and hold the secret to a successful γ-secretase AD clinical trial.
I do not endorse the view that the trial was started prematurely. One has to do the best with what is on hand for such a debilitating disease. However, I do fully endorse the call for more basic research on γ-secretase. In my view, the most robust and informative approach would be from a collective perspective of functionally related genes, unbiased by any dogmas or models. Dogma is a hindrance. Models are good for designing experiments and interpreting results, but also restrict freedom and delay discovery. Many of my journeys were prompted by totally unexpected findings. Some led nowhere but others led to exciting insights. For example, a “boring” experiment to determine how long the effect of a temperature-sensitive Notch mutation lasts led to the exciting discovery of CREB ultradian oscillation in memory formation (Zhang et al., 2013). Further research on γ-secretase could exploit the empirical tools and relative simplicity of the Drosophila model system that has hitherto provided a sound framework for initiating studies in mammalian systems.
Even if γ-secretase ultimately proves to be a bad drug target, I believe an unbiased research approach would lead to unexpected good targets for AD. I predict that even these good targets would somehow connect to γ-secretase activity. However, I am confident that the knowledge acquired in the discovery process would point to ways and means for severing that connection. As an illustration, if I were to look for inhibitors of Notch, I would start a detailed analysis of the naturally produced dominant-negative molecules that are likely to produce fewer side effects. If I were to look for a better way to deliver γ-secretase inhibitor to humans, I would start by identifying the peaks and troughs of Notch activities and explore the effect of drug delivery at peak or trough times depending on the effect desired (note that ultradian oscillation of Notch activity is reported in vertebrates and known to be important for somitogenesis).
I suspect most scientists agree with De Strooper’s call for more basic and applied research on γ-secretase. I do, too. But I disagree on the means for achieving that goal. De Strooper argues for more money from funding agencies and more investment by companies. In my view, money is not the limiting resource for sound research on the subject, and companies have to make business decisions not charitable ones.
References:
Zhang J, Little CJ, Tremmel DM, Yin JC, Wesley CS. Notch-Inducible Hyperphosphorylated CREB and Its Ultradian Oscillation in Long-Term Memory Formation. J Neurosci. 2013 Jul 31;33(31):12825-34. PubMed.
View all comments by Cedric Wesleyat Forum Pharmaceuticals
Arkuda Therapeutics
Bart De Strooper makes a passionate plea to continue exploring γ-secretase as a molecular target for Alzheimer’s disease. In general we agree with his assessment that γ-secretas e has received a bad reputation because of the failures of a few prominent clinical trials, namely semagacestat and avagacestat, which were supposed to test the amyloid hypothesis by inhibiting this enzyme complex. Where we disagree slightly is on the emphasis De Strooper puts on renewed testing of truly substrate-specific γ-secretase inhibitors (GSIs). We would favor γ-secretase modulators (GSM) in a clinical setting. Further research into the exact molecular mechanism of the γ-secretase heteromeric enzyme complex should enable the discovery of better GSI molecules. That effort will most likely also help in the discovery and possibly interpretation for GSMs. The key is to test the best possible approach (the right mechanism) in expensive clinical trials.
γ-Secretase as a Target and the Amyloid Hypothesis
The amyloid hypothesis in its original form drew from the observation that the Aβ42:Aβ40 ratio was increased in patients with familial Alzheimer’s disease (FAD). It proposed that increased production of Aβ42 and its accumulation might lead to a number of downstream effects, which would culminate in neuronal dysfunction and ultimately dementia. The inhibition of a key enzyme responsible for the production of the various Aβ peptides appeared attractive at first. However, careful analysis of the presenilin 1 (PS1) mutations found in FAD cases has led to the suggestion that the molecular defect caused by these mutants renders the γ-secretase complex defective in the processing of APP into the various shorter Aβ peptides, thus leading to the increased Aβ42:Aβ40 ratio. This has been elegantly demonstrated by Masa Okochi and colleagues from Osaka University in a report where they show that presenilin mutations decrease the catalytic activity of the γ-secretase complex (Okochi et al., 2012).
Based on these findings, we should probably reconsider whether inhibition of γ-secretase is the best option if we target this enzyme complex. We thus agree with De Strooper that we should be more careful in stating that γ-secretase as a molecular target is “out.” Based on his own work (Chávez-Gutiérrez et al., 2012) and the work of Okochi and colleagues mentioned above, we might want to consider GSMs as a therapeutic approach to correct the defect caused by presenilin mutations in early onset FAD. The challenge will be how to translate a potential future proof-of-mechanism in the clinic in a Phase 1b/2a study (i.e., movement of the biomarker Aβ in the CSF and not plasma) to a relevant clinical endpoint in FAD and eventually into sporadic AD (see below).
Semagecestat and Lessons
As several contributors already mentioned, target engagement is critically important in initial proof-of-mechanism studies in early drug development. For semagacestat, it seems that target engagement was achieved at the higher doses but the issue was that compound exposure only covered target engagement for 12 hours (Bateman et al., 2009). We can only speculate that Lilly might have had additional data which increased their confidence that Aβ was significantly reduced over 24 hours. As others have pointed out, the chosen dose in Phase 3 was lower and probably did not result in sufficient exposure to lower Aβ for a long enough period of time. One important lesson from this experience is that any trial going forward needs to assure that the targeted reduction of the biomarker of interest (Aβ total, Aβ42, etc.) is achieved over the whole course of the day, not just 12 hours.
Obviously, this was one of many possible reasons why the interpretation of this trial was more complicated than anticipated. Another potential issue specific to semagacestat appears to be the susceptibility to active efflux at the blood-brain barrier (BBB), as described by Lu et al. (2011). While this undesirable property does not necessarily prevent a compound from development, it could add additional variability to the brain exposure in an already highly variable patient population in the Phase 3 trial. We therefore would not attempt to interpret the semagacestat clinical data further without additional experimental evidence.
Biomarker, Timing of Intervention, Patient Population and Clinical Endpoints
In addition, the timing of intervention remains a huge challenge. As Eric Karran, Marc Mercken, and De Strooper have nicely explained in their 2011 review, early intervention provides the highest chance of success when it comes to Aβ-lowering approaches. Unless we assume that the acute production of newly formed monomeric Aβ42/Aβ43 provides a new “toxic insult” to a brain already full of amyloid deposits, it is safe to say that we might have to treat before the clinical diagnosis. But how do you assess clinical and functional decline in a patient population that won’t progress rapidly? The current available assessment tools appear to be insufficient for pre-symptomatic or even early stage diagnosis. One approach is to design conversion trials (e.g., pre-symptomatic to mild cognitive impairment, MCI). Given the slow decline in the early stages, these trials would require huge numbers of patients and would by definition be very long to allow for enough statistical power. Another alternative would be a more focused approach: Test the agents in a population where we understand better when and why patients convert to AD. That is where initiatives like DIAN are the most logical path forward. Indeed, several companies are choosing this path with their therapeutics. But here we are talking about few and precious patients since FAD patients are a small fraction of the AD population.
Our field will be given only a few more reasonable chances to show some clinical success in testing the amyloid hypothesis. Some might argue that one more antibody failure combined with lack of efficacy of the ongoing BACEi trials will cause a shutdown of most, if not all, investment in targets aimed at amyloid reduction. This is why we believe that an intervention with a GSM in pre-symptomatic FAD patients has the best scientific rationale and, with the right molecule, the highest probability of success. GSMs deserve priority in our mind over new, more substrate-specific GSIs in that hypothesis testing scenario.
In summary, we think γ-secretase research should be supported to allow the field to understand this enzyme complex better. Even though the GSM field has yet to deliver a promising clinical candidate (lack of good drug properties being the major hurdle), we think that the priority on the initial clinical development should lie with GSM molecules for the reasons mentioned.
References:
Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M. γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013 Jan 31;3(1):42-51. PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009 Jul;66(1):48-54. PubMed.
Lu Y, Zhang L, Nolan CE, Becker SL, Atchison K, Robshaw AE, Pustilnik LR, Osgood SM, Miller EH, Stepan AF, Subramanyam C, Efremov I, Hallgren AJ, Riddell D. Quantitative pharmacokinetic/pharmacodynamic analyses suggest that the 129/SVE mouse is a suitable preclinical pharmacology model for identifying small-molecule γ-secretase inhibitors. J Pharmacol Exp Ther. 2011 Dec;339(3):922-34. PubMed.
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011 Sep;10(9):698-712. PubMed.
View all comments by Jean-Francois BlainLilly Corporate Center
Eli Lilly and Company
Eli Lilly and Company
Eli Lilly and Company
The semagacestat development team at Eli Lilly and Company has noted the paper from Dr. De Strooper with great interest. Professor De Strooper makes a number of excellent points with regard to drug development broadly. He also raises a number of questions specific to the non-clinical and clinical development of semagacestat. We appreciate the opportunity provided by Alzforum to advance the field of AD therapeutics by replying to some of these points in detail.
In summary, we agree that the pharmaceutical industry should be persuaded to sustain its interest in many types of Aβ or amyloid targeting therapies, including γ-secretase inhibitors. While we now know that the γ-secretase complex consists of four subunits (presenilin, nicastrin, presenilin enhancer 2, and anterior pharynx 1) encoded by four different genes (PSEN, NCT, PEN2, and APH1) (De Strooper, 2003), even the relationship between presenilin and γ-secretase was found after development of semagacestat had begun. In our view, taken together, the data would suggest that the cognitive worsening seen in the IDENTITY trial was most likely due to inhibition of cleavage of a γ-secretase substrate other than APP. Thus, perhaps through targeting (or not targeting) one particular subunit of γ-secretase, selective inhibition of APP cleavage could be achieved, or γ-secretase modulation could obviate the cognitive worsening seen in IDENTITY. During the past decade, much has been learned about the γ-secretase complex, and much has been learned from the clinical results obtained from trials using semagacestat and other similar γ-secretase inhibitors. The field can build on these learnings and continue to work toward finding safe and effective treatments for AD that target the γ-secretase complex.
References:
Greenberg BD, Carrillo MC, Ryan JM, Gold M, Gallagher K, Grundman M, Berman RM, Ashwood T, Siemers ER. Improving Alzheimer's disease phase II clinical trials. Alzheimers Dement. 2013 Jan;9(1):39-49. PubMed.
Siemers E. Comment: Is Alzheimer’s disease drug development broken? What must be improved . J Prev Alz Dis. 2014;1:8-10.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, , Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341-50. PubMed.
Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014 Aug;76(2):185-205. Epub 2014 Jul 2 PubMed.
Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009 Jul;66(1):48-54. PubMed.
Portelius E, Dean RA, Gustavsson MK, Andreasson U, Zetterberg H, Siemers E, Blennow K. A novel Abeta isoform pattern in CSF reflects gamma-secretase inhibition in Alzheimer disease. Alzheimers Res Ther. 2010;2(2):7. PubMed.
Henley DB, May PC, Dean RA, Siemers ER. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin Pharmacother. 2009 Jul;10(10):1657-64. PubMed.
Gitter BD, Czilli DL, Li W, Dieckman DK, Bender MH, Nissen JS, Mabry TE, Yin T, Boggs LN, McClure DB, Little SP, Johnstone EM, Audia JE, May PC, Hyslop PA. Stereoselective inhibition of amyloid beta peptide secretion by LY450139, a novel functional gamma secretase inhibitor. Neurobiol Aging. 2004; 25(Suppl 2):S571.
Henley DB, Sundell KL, Sethuraman G, Dowsett SA, May PC. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr Med Res Opin. 2014 Oct;30(10):2021-32. Epub 2014 Jul 14 PubMed.
Lanz TA, Karmilowicz MJ, Wood KM, Pozdnyakov N, Du P, Piotrowski MA, Brown TM, Nolan CE, Richter KE, Finley JE, Fei Q, Ebbinghaus CF, Chen YL, Spracklin DK, Tate B, Geoghegan KF, Lau LF, Auperin DD, Schachter JB. Concentration-dependent modulation of amyloid-beta in vivo and in vitro using the gamma-secretase inhibitor, LY-450139. J Pharmacol Exp Ther. 2006 Nov;319(2):924-33. PubMed.
May PC, Yang Z, Li WY, Hyslop PA, Siemers E, Boggs LN. Multicompartmental pharmaco-dynamic assessment of the functional gamma-secretase inhibitor LY450139 in PDAPP transgenic mice and non-transgenic mice. . Neurobiol Aging. 2004;25(Suppl 25):S65.
Boggs LN, Fuson KS, Gitter BD, Czilli DL, Hyslop PA, Bender MH, Li W, Audia JE, Nissen JS, Mabry TE, Ni B, Su Y, May PC. In vivo characterization of LY450139, a novel, stereoselective, functional gamma-secretase inhibitor. Neurobiol Aging. 2004 Jul;25(Suppl 2):S218.
Hyslop PA, May PC, Audia JE, Calligaro DO, McMillian CL, Garner CO, Cramer JW, Gitter BD, Porter WJ, Nissen JS, Mabry TE, Bender MH. Reduction in A-beta(1-40) and A-beta(1-42) in CSF and plasma in the beagle dog following acute oral dosing of the gamma secretase inhibitor, LY450139. Neurobiol Aging. 2004 Jul;25(Suppl 2):S147.
Ness DK, Boggs LN, Hepburn DL, Gitter B, Long GG, May PC, Piroozi KS, Schafer KA, Yang Z. Reduced β-amyloid burden, increased C-99 concentrations and evaluation of neuropathology in the brains of PDAPP mice given LY450139 dihydrate daily by gavage for 5 months. Neurobiol Aging. 2004 Jul; 25(Suppl 2):S238-9.
Lanz TA, Hosley JD, Adams WJ, Merchant KM. Studies of Abeta pharmacodynamics in the brain, cerebrospinal fluid, and plasma in young (plaque-free) Tg2576 mice using the gamma-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]az. J Pharmacol Exp Ther. 2004 Apr;309(1):49-55. PubMed.
De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003 Apr 10;38(1):9-12. PubMed.
View all comments by Karen SundellUniversity of California, Irvine
The clinical trial of semagacestat is fascinating because it reported the first disease-modifying drug for Alzheimer’s. The critical question is why the drug made the patients cognitively worse. A number of people have pointed to an off-target effect on other γ-secretase substrates, like Notch. While these and other interpretations are reasonable, they are not the simplest explanation. The simplest explanation is that we are thinking about the mechanistic relationship between the γ-secretase processing of APP-CTF and pathogenesis backwards. The common view of the amyloid hypothesis that formed the basis for the development of semagacestat holds that the secretion of short, soluble Aβ species ending at residues 40 or 42 is pathological. The inverse mechanism is that the intraneuronal retention of insoluble, long Aβ species is pathological. Both views are supported by the human genetics underlying the amyloid hypothesis, but the semagacestat results suggest that the latter view is more likely. γ-secretase initially cleaves APP-CTF after residue 49 of the Aβ sequence, and then processively trims this long Aβ species until it results in the soluble products secreted by the cell. It is known that FAD mutations in presenilins interfere with this exopeptidase processivity, resulting in an increase in the steady-state levels of long Aβ species (Fernandez et al., 2014), that have high aggregation potential unless they are degraded. The γ-secretase inhibitor DAPT also increases the levels of long Aβ species (Yagishita et al., 2006) thus providing a facile explanation for why semagacestat made the patients cognitively worse: It does the same thing that FAD mutations in presenilin do. They both increase the levels of long Aβ species that may aggregate inside the neuron. We have recently published a more detailed view of intraneuronal amyloid aggregation and an alternative view of the amyloid hypothesis (Pensalfini et al., 2014).
References:
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
Yagishita S, Morishima-Kawashima M, Tanimura Y, Ishiura S, Ihara Y. DAPT-induced intracellular accumulations of longer amyloid beta-proteins: further implications for the mechanism of intramembrane cleavage by gamma-secretase. Biochemistry. 2006 Mar 28;45(12):3952-60. PubMed.
Pensalfini A, Albay R 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, Kawas CH, Glabe CG. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol Dis. 2014 Nov;71:53-61. Epub 2014 Aug 1 PubMed.
View all comments by Charles GlabeSloan-Kettering Institute
Bart de Strooper conducted a comprehensive analysis of clinical studies of Semagacestat based on published data and made a convincing argument that the failed clinical trial does not disqualify γ-secretase as a target for AD drug development. We should focus on lessons learned from failed clinical trials and develop a better understanding of γ-secretase structure and function under physiological and pathophysiological conditions, which would provide a molecular basis for development of effective and safe γ-secretase-based therapy for AD treatment. The most surprising outcome of the clinical trial of Semagacestat was the worsening of memory in patients (Doody et al., 2013). The other major adverse effect of the trial was the increased risk of skin cancer, which likely resulted from inhibition of Notch signaling by Semagacestat (Xia et al., 2001; Nicolas et al., 2003). While the Notch-associated side effects are somewhat better understood, the mechanism of cognitive decline is elusive. It may be informative to compare clinical studies of the γ-secretase inhibitor Avagacestat (Coric et al., 2012).
With Avagacestat, although the major adverse effects included predominant gastrointestinal and dermatologic complications, the 100 mg and 125 mg dose arms also led to a trend for cognitive worsening (Coric et al., 2012), suggesting that both compounds may share a common mechanism of toxicity. It is noteworthy to point out that although many proteins have been reported to be substrates of γ-secretase (Haapasalo and Kovacs, 2011), it is not known how many of these are truly cleaved by γ-secretase in vivo. This is an important question to answer in order to predict which substrates are most likely to cause toxicity when γ-secretase is inhibited. Avagacestat was reported as a Notch sparing inhibitor (Gillman et al., 2010). However, the specificity of this compound is questionable (Chavez-Gutierrez et al., 2012; Crump et al., 2012).
Likewise, the selectivity of Semagacestat is in debate, as mentioned above. Clearly, the AD research community from academia to industry should coordinate efforts to develop methodologies and consensus standards for evaluating the potency and selectivity of molecules that target γ-secretase. These clinical studies call for the development of γ-secretase modulators or other disease modifying agents that lessen γ-secretase activity for the production of Aβ42 without affecting the overall processing and function of other γ-secretase substrates (Weggen et al., 2001; Crump et al., 2013). γ-Secretase is composed of four obligatory subunits (Presenilin, Nicastrin, Pen2 and Aph-1) (De Strooper, 2003; Edbauer et al., 2003; Takasugi et al., 2003) that form multiple enzyme complexes, but it is also modulated by an array of regulatory subunits (Gertsik et al., 2014), such as GSAP (He et al., 2010) and HIF1α (Villa et al., 2014). It is important to investigate how these non-essential subunits modulate γ-secretase activity in AD and affect the potency and specificity of small molecules. We appreciate now that the regulation of γ-secretase is highly complex and may be playing an important role in disease states.
Taken together, as indicated by Bart de Strooper, failures in the Semagacestat clinical trial can be reasonably explained and offer valuable lessons for further development of γ-secretase-targeted AD therapies. They do not disprove γ-secretase as a valid drug target. More importantly, the development of safe and effective therapies requires a comprehensive understanding of the target – γ-secretase. We have witnessed recent success in the development of cancer immunotherapy after decades of disappointments (Mellman et al., 2011). There is hope that safe and effective AD therapies will be developed in the near future.
References:
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Crump CJ, Castro SV, Wang F, Pozdnyakov N, Ballard TE, Sisodia SS, Bales KR, Johnson DS, Li YM. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry. 2012 Sep 18;51(37):7209-11. PubMed.
Crump CJ, Johnson DS, Li YM. Development and Mechanism of γ-Secretase Modulators for Alzheimer's Disease. Biochemistry. 2013 May 2; PubMed.
De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003 Apr 10;38(1):9-12. PubMed.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, , Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341-50. PubMed.
Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003 May;5(5):486-8. PubMed.
Gertsik N, Chiu D, Li YM. Complex regulation of γ-secretase: from obligatory to modulatory subunits. Front Aging Neurosci. 2014;6:342. Epub 2015 Jan 6 PubMed.
Gillman KW Jr, Starrett JE Jr, Parker MF, Xie K, Bronson JJ, Marcin LR, McElhone KE, Bergstrom CP, Mate RA, Williams R, Meredith JE, Burton CR, Barten DM, Toyn JH, Roberts SB, Lentz KA, Houston JG, Zaczek R, Albright CF, Decicco CP, Macor JE, Olson RE. Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable γ-Secretase Inhibitor. ACS Med Chem Lett. 2010 Jun 10;1(3):120-4. Epub 2010 Mar 22 PubMed.
Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, Soininen H, Thein S, Shiovitz T, Pilcher G, Colby S, Rollin L, Dockens R, Pachai C, Portelius E, Andreasson U, Blennow K, Soares H, Albright C, Feldman HH, Berman RM. Safety and Tolerability of the γ-Secretase Inhibitor Avagacestat in a Phase 2 Study of Mild to Moderate Alzheimer Disease. Arch Neurol. 2012 Aug 13;:1-12. PubMed.
Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. J Alzheimers Dis. 2011;25(1):3-28. PubMed.
He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein is a therapeutic target for Alzheimer's disease. Nature. 2010 Sep 2;467(7311):95-8. PubMed.
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003 Mar 27;422(6930):438-41. PubMed.
Villa JC, Chiu D, Brandes AH, Escorcia FE, Villa CH, Maguire WF, Hu CJ, de Stanchina E, Simon MC, Sisodia SS, Scheinberg DA, Li YM. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014 Aug 21;8(4):1077-92. Epub 2014 Aug 14 PubMed.
Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10863-8. PubMed.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011 Dec 21;480(7378):480-9. PubMed.
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003 Mar;33(3):416-21. Epub 2003 Feb 18 PubMed.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001 Nov 8;414(6860):212-6. PubMed.
UK Dementia Research Institute@UCL and VIB@KuLeuven
I would like to thank all contributors to the discussion for sharing really interesting thoughts. I also strongly appreciate the participation of industry and their constructive input. This type of interaction is very helpful and shows how far the field has moved on from the disastrous debates in the AD field years ago. From my side I want to address briefly four issues.
1. I agree that activators of the enzyme, e.g. activating its carboxypeptidase-like ‘trimming’ function, is an interesting way to go and in agreement with our own publications (De Strooper, 2007, Chávez-Gutiérrez et al., 2012). Such an approach would be particularly strong in combination with a BACE1 inhibitor, as this would act both on quantity and quality of the Aβ production (Strömberg et al., 2015) and impact intracellular Aβ, as well.
2. An important lesson of the Semagacestat trial is that pharmacodynamics count. The trial dose resulted in enormous peaks of inhibitor, i.e. > 2,000 ng/ml in blood (Yi et al., 2010) and close to 200 ng/ml in CSF (Bateman et al., 2009) , respectively, to be compared with an IC50 of Semagacestat in cells of about 6 ng/ml. This results in periods of complete block of γ-secretases, while, as discussed, even a short complete block of Notch signaling is probably sufficient to precipitate important side effects.
Splitting the 140 mg dose into two doses (which was apparently tested by the Lilly investigators) does not necessarily address this problem and may actually worsen the situation because you create, instead of one, two peaks of relative high drug exposure over 24 hours. From a side effect perspective, it is clearly better to inhibit γ-secretase in a continuous way for 40 percent over the day than to hit strongly at 100 percent, followed by drug-free periods either once or twice a day. As Semagacestat has a short half-life in blood and CSF (about 2.5 hours), this would require multiple dosing over 24 hours to build up a steady-state concentration that remains below the threshold for complete Notch inhibition. More phase II trials to determine the optimal pharmacokinetics for the compound would have been useful to test whether such a window existed. Likely such a steady-state regimen would also result in clear effects on Aβ levels in the CSF. The conclusion is not that Semagacestat should be further clinically developed, but that we need more, smaller trials to evaluate and probe the potential of new drugs before going into phase III and making definitive conclusions.
3. This brings me to a more philosophical note: as mentioned by several discussants, drug development for AD goes currently very rapidly to phase III trials. I think this is an unsustainable approach, especially since there are still many uncertainties about the drug targets, their physiology in the elderly, and about the different subgroups in Alzheimer disease. The phase III trials take too long and are too expensive. They are essentially all-or-nothing experiments, making it difficult to retest a failed compound in a different way. That is the main reason why we need more exploratory trials, testing the effects of the drugs first on relevant biomarkers. This would allow identifying pharmacodynamic problems upfront, defining the best patient subgroup, but also determining whether the target (in case of Semagacestat: Aβ lowering in the brain) can be hit in a convincing way.
Time pressure because of the patent life cycle is a major problem in the decision-making process. Non-orthodox initiatives to stimulate drug development in the AD field could be considered. For example, would prolongation of patent protection for drugs coming from such trials be helpful? One could compensate partially for the extra time that clinical trials in the AD field take compared to other disease areas.
4. Finally, the granting bodies and their referees have hopefully read this discussion and noticed the clear support for further basic research on these important drug targets both from academy and industry. The case of γ-secretases is not closed.
References:
Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009 Jul;66(1):48-54. PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):141-6. PubMed.
Strömberg K, Eketjäll S, Georgievska B, Tunblad K, Eliason K, Olsson F, Radesäter AC, Klintenberg R, Arvidsson PI, von Berg S, Fälting J, Cowburn RF, Dabrowski M. Combining an amyloid-beta (Aβ) cleaving enzyme inhibitor with a γ-secretase modulator results in an additive reduction of Aβ production. FEBS J. 2015 Jan;282(1):65-73. Epub 2014 Nov 7 PubMed.
Yi P, Hadden C, Kulanthaivel P, Calvert N, Annes W, Brown T, Barbuch RJ, Chaudhary A, Ayan-Oshodi MA, Ring BJ. Disposition and metabolism of semagacestat, a {gamma}-secretase inhibitor, in humans. Drug Metab Dispos. 2010 Apr;38(4):554-65. Epub 2010 Jan 14 PubMed.
Case Western Reserve University
“Don't waste clean thinking on a dirty enzyme.”
In this thread, the question of whether γ-secretase should remain a target has generated a widespread response from experts in the field covering nearly every investigative arena from basic science, translational science, biomarkers, clinical trials, and the pharmaceutical industry. Together, they address different aspects of the failed semagacestat trial and most commentators conclude that “… clinical investigation of γ-secretase as a therapeutic target should continue.” However, I think this conversation has missed an important point.
What the field urgently needs is a treatment that works for Alzheimer’s disease patients, not to tweak around the edges of an approach that has not worked. If anything, semagacestat pharmacodynamics, the differential effects of γ-secretase inhibition on Notch and APP, the paradoxical increase in Aβ at low inhibitor levels, etc., collectively suggest that one cannot devise a more difficult approach to reduce Aβ levels than γ-secretase inhibition. It is somewhat surprising that inhibition of Aβ generation is still considered a viable approach, even though AD patients exhibit decreased clearance of Aβ from brain parenchyma rather than excess generation (Mawuenyega et al., 2010). Even if one believes in the amyloid hypothesis, and even if reducing Aβ levels were to result in improved cognition (notwithstanding contrary evidence, e.g., Giacobini and Gold, 2013), one must see that sustained inhibition/modulation of γ-secretase by chemical means over a period of years has to be the least optimal way to treat AD patients.
The quote above by the great biochemist Efraim Racker alluded to wasting resources (both intellectual and financial) on studying impure, contaminated enzyme preparations. I think γ-secretase, as a drug target, can be viewed as a “dirty enzyme.” It has more than 70 substrates and many of them regulate critical physiological events. Prolonged use of any chemical inhibitor, which would be necessary in the prevention trials paradigm, is bound to affect these non-amyloid pathways and cause side effects. Termination of the BACE inhibitor BI1181181 trial further proves this point (see Feb 2015 news). Even the highly selective statins, which inhibit an enzyme with only one substrate, have considerable side effects.
γ-secretase is fascinating and the field should continue to investigate it as an enzyme that regulates important cellular pathways, but not as a drug target for AD. The limited resources of the NIH and Alzheimer’s Association would be better targeted at therapies that have already shown efficacy against AD and dementia, namely physical exercise (Chapman et al., 2013). Emerging data show that regular physical exercise is the only proven therapy against cognitive loss due to aging and/or AD. In controlled clinical trials, regular physical exercise was shown to be effective not only in preventing (Schlosser Covell et al., 2015), but also in reversing cognitive decline in elderly patients with MCI (Nagamatsu et al., 2013). We do not precisely understand the cellular and molecular mechanisms underlying the beneficial effects of exercise on brain function, and allocating resources toward their investigation may turn out to be more beneficial than continuing to pursue strategies that, despite repeated attempts, have yet to yield positive results.
References:
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010 Dec 24;330(6012):1774. PubMed.
Giacobini E, Gold G. Alzheimer disease therapy--moving from amyloid-β to tau. Nat Rev Neurol. 2013 Dec;9(12):677-86. Epub 2013 Nov 12 PubMed.
Chapman SB, Aslan S, Spence JS, Defina LF, Keebler MW, Didehbani N, Lu H. Shorter term aerobic exercise improves brain, cognition, and cardiovascular fitness in aging. Front Aging Neurosci. 2013;5:75. Epub 2013 Nov 12 PubMed.
Schlosser Covell GE, Hoffman-Snyder CR, Wellik KE, Woodruff BK, Geda YE, Caselli RJ, Demaerschalk BM, Wingerchuk DM. Physical activity level and future risk of mild cognitive impairment or dementia: a critically appraised topic. Neurologist. 2015 Feb;19(3):89-91. PubMed.
Nagamatsu LS, Chan A, Davis JC, Beattie BL, Graf P, Voss MW, Sharma D, Liu-Ambrose T. Physical activity improves verbal and spatial memory in older adults with probable mild cognitive impairment: a 6-month randomized controlled trial. J Aging Res. 2013;2013:861893. Epub 2013 Feb 24 PubMed.
View all comments by Sanjay PimplikarMake a Comment
To make a comment you must login or register.