Research Brief: Driven to Diffraction—X-rays Show APP Dimerization
Quick Links
There is more to amyloid precursor protein (APP) than the peptide for which it is named. At 40-42 amino acids, amyloid-β (Aβ) may attract the most attention, but the full protein is almost 20 times as large, with most of it protruding from the cell surface. While the extracellular expanse of APP may be its business end, relatively little is understood about what it does. Some evidence suggests that the protein forms dimers, which could be important for function, and a paper in this week’s PNAS supports that idea. Researchers led by Manuel Than at the Leibniz Institute for Age Research-Fritz Lipmann Institute, Jena, Germany, crystallized one part of the APP extracellular domain. Analysis of the crystal structure predicts that it, and by extrapolation APP itself, should form a dimer in the presence of heparin.
“It is now very probable that cellular signaling of APP indeed uses the well-established principle of receptor dimerization, and in the future, other biomolecules, in addition to heparin/heparan sulfate, may be found to influence the APP monomer/dimer equilibrium,” noted Mathias Gralle, Max Planck Institute for Evolutionary Anthropology, Leipzig, Germany, in an e-mail to ARF (see full comment below). Gralle was not involved in the work but has studied the three-dimensional structure of APP in solution (see Gralle et al., 2006).
Transmembrane proteins such as APP are notoriously difficult to crystallize. Researchers generally get around this by studying smaller pieces rather than the whole protein. First author Sven Dahms and colleagues focused on the E1 N-terminal region of APP comprising copper binding (CuBD) and growth factor-like domains (GFLDs). They obtained enough of the protein fragment for crystallization by expressing it in and purifying it from Escherichia coli. Analysis of the crystals showed that the two domains rigidly interact to form one folding unit (see image below). “The solved structure could have a strong impact on function, because it indicates the two domains cannot work independently,” Than told ARF. How function might be affected by this rigid structure remains to be seen.
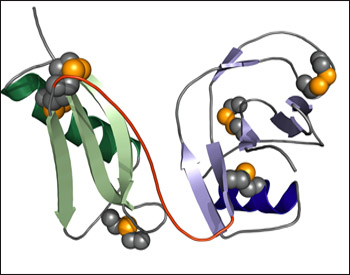
3-D Structure of the E1 N-terminal Domain of APP
The CuBD (green) and the GFLD (blue) form distinct folds connected by an interdomain linker (red). Dark colors represent α helices and lighter colors β-sheets. Disulphide bonds are shown as gold spheres. Image credit: Manuel Than, Leibniz Institute for Age Research-Fritz Lipmann Institute
The analysis also indicates that two E1 fragments can come together to form a dimer in the presence of heparin, which is known to bind APP. The protein dimer interface comprises positively charged amino acid side chains that are held in place by the negatively charged heparin. This would likely affect the whole structure of APP because “if E1 comes together, then the whole molecule must come together,” said Than.
Interestingly, the researchers found that the stability of the rigid interdomain structure of the E1 monomer is pH dependent, being tighter at slightly acidic pH (~5.0). The researchers suggest that APP could form distinct conformations in different cellular and extracellular milieus.—Tom Fagan
References
Paper Citations
- Gralle M, Oliveira CL, Guerreiro LH, McKinstry WJ, Galatis D, Masters CL, Cappai R, Parker MW, Ramos CH, Torriani I, Ferreira ST. Solution conformation and heparin-induced dimerization of the full-length extracellular domain of the human amyloid precursor protein. J Mol Biol. 2006 Mar 24;357(2):493-508. PubMed.
Further Reading
Primary Papers
- Dahms SO, Hoefgen S, Roeser D, Schlott B, Gührs KH, Than ME. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2010 Mar 23;107(12):5381-6. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Universidade Federal do Rio de Janeiro
While there is a wealth of studies proposing different roles for APP in
health and disease, mechanistic explanations for the functions of APP
are still not as reliable as one would wish. Structural detail seems the
way forward to sift through the proposed mechanisms and agree on the most
plausible ones.
A few years ago, several fragments of APP were solved
independently at atomic resolution, but it seemed impossible to get the
same level of resolution for the full-length protein. This would be
a serious problem for studying regulation and cooperation between its
different binding partners. Now Dahms et al. prove that it is indeed
possible to study the interactions between two domains of APP, the
N-terminal growth factor-like domain and the copper-binding domain, and
derive new mechanistical insight from this. This opens up the
possibility of further progress towards a high-resolution understanding
of full-length APP.
One major conclusion of Dahm et al. is that heparin indeed induces
dimerization of their object of study, the two most N-terminal domains
of APP. This role of heparin/heparan sulfate had already been suggested
for both full-length extracellular APP (sAPP) and for membrane-bound APP
in cells, but Dahm et al. add atomic-level detail and thermodynamic data
that were impossible to obtain for the larger, more complex sAPP and
APP. It is now very probable that cellular signaling of APP indeed uses
the well-established principle of receptor dimerization,
and in the future other biomolecules, in addition to heparin/heparan
sulfate, may be found to influence the APP monomer/dimer equilibrium.
Furthermore, Dahm et al. show that the two N-terminal domains form a
tight interface at acidic pH in the crystal (and probably in solution),
which has been evolutionarily conserved. The combination of structural
and evolutionary data suggest strongly that this interaction between the
two domains has biological importance; the most parsimonious explanation
would be that while invertebrate APP-like proteins and vertebrate APLP1
contain independent domains, the common precursor of vertebrate APP and
APLP2 acquired an interaction between them.
Dahm et al. also suggest, using limited proteolysis data, that the domain interface is still
present, albeit in a weaker form, at higher pH, which is in conflict
with earlier small-angle X-ray scattering and analytical
ultracentrifugation data on the same fragment of APP. The important
point may be that the interaction between the two domains is weak and
subject to multiple influences and regulators, such as local pH, copper,
and zinc ions. This is an exciting first glimpse at how APP manages to
integrate the actions of its multiple binding partners and transmit
different signals in different cellular compartments.
Make a Comment
To make a comment you must login or register.