Not All About Dendrites: Presynaptic Tau Harms Plasticity, Too
Quick Links
Many studies link tau to synaptic dysfunction and memory loss in Alzheimer’s disease and other dementias. But how does tau do its dirty work? Most previous research focused on the harmful effects of tau that strays into dendrites and acts on postsynapses there. In the April 3 Acta Neuropathologica Communications, researchers led by Eva-Maria and Eckhard Mandelkow at the German Center for Neurodegenerative Diseases, Bonn, turn the spotlight on presynaptic tau. Because tau is an axonal protein, it is likely to occupy presynapses earlier than postsynapses, the Mandelkows told Alzforum. In mouse tauopathy models, small tau aggregates in hippocampal presynapses lowered calcium influx, impaired plasticity, and precipitated the loss of synapses. Inhibitors of aggregation preserved synaptic structure, strengthening the case for aggregated tau as the toxic entity. It is not yet clear whether presynaptic deficits precede postsynaptic problems, or vice versa.
“This is one of the first papers to explore the function of presynaptic tau,” noted Amy Pooler at the Nestlé Institute of Health Sciences in Lausanne, Switzerland. She praised the thoroughness of the study, which ranged from in vivo to slice and cell culture experiments. Others agreed. “The paper represents a typically excellent body of work from the Mandelkow lab,” George Bloom at the University of Virginia, Charlottesville, wrote to Alzforum.
Previous studies by the Mandelkows and others found that in Alzheimer’s models, tau, ordinarily a microtubule-binding protein, wanders off into dendrites, where it contributes to excitotoxicity caused by Aβ (see Jul 2010 conference news; Sep 2010 news; Jan 2011 news). These experiments left unclear what form of tau harms synapses. To pin this down, the Mandelkows generated mouse models that express either a form of tau that aggregates easily, called TauRDΔ, or one that never does. Only TauRDΔ mice developed Alzheimer’s-like pathology and memory problems, they reported (see Jan 2008 news; Feb 2011 news).
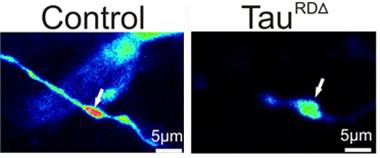
Calcium Deficit.
In control animals (left panel), boutons flood with calcium (red) after depolarization, but little calcium enters boutons in pro-aggregant tau mice (right panel). [Image courtesy of Decker et al., 2015, BioMed Central.]
In the current study, the Mandelkows used these mice to investigate the presynaptic effects of tau aggregates. Presynapses are generally harder to study than postsynapses because they are harder to identify. Joint first authors Jochen Martin Decker and Lars Krüger analyzed mossy fibers, axons that extend from neurons in the granule cell layer of the dentate gyrus to contact dendrites of pyramidal cells in area CA3 of the hippocampus. Mossy fibers form one of the largest presynapses in the brain, enabling detailed studies of its structure and function. These excitatory synapses underpin memory storage and retrieval, and their loss correlates with the onset of severe memory deficits in AD patients. In TauRDΔ mice, the CA3 region develops the most pronounced pathology, Eva-Maria Mandelkow said.
These synapses functioned normally in young TauRDΔ animals, but by 13 months of age, the mossy fiber boutons contained hyperphosphorylated, oligomeric tau and looked abnormal. The boutons swelled by about 50 percent, but contained less than half the number of synaptic vesicles of controls. Moreover, these synapses transmitted weakly and were less plastic than those in wild-type mice and in animals expressing non-aggregating tau. Why might this be? Knowing that calcium must enter presynapses for normal plasticity to occur, the authors measured calcium flux in the mossy fibers in hippocampal slices. Slice cultures came from young mice but developed pathology after 10 days in vitro. In tissue from TauRDΔ mice, about one-third less calcium flooded into boutons after depolarization compared to control slices. The authors also saw a 20 percent drop in spine density in the CA3 region. Similarly, calcium influx slowed in primary neurons and cell lines expressing the aggregating form of tau, dropping by two-thirds in cells with visible tau deposits, again fingering tau aggregates as the culprits.
Could blocking tau aggregation save synapses? When the authors treated brain slices with an aggregation inhibitor, it prevented the ballooning of boutons and the loss of spines. These studies had to be done in slice culture because most tau aggregation inhibitors do not penetrate the blood-brain barrier well, Eva-Maria Mandelkow said.
Tau aggregation is not the whole picture, however. The authors also examined tau knockout mice. While these animals had normal synaptic plasticity, they did grow enlarged boutons. Their synapses also transmit weakly, just like the animals expressing the aggregation-prone tau. Bloom was intrigued by the finding that both aggregated tau and a lack of tau perturb synaptic function. “Together, these results raise the possibility that tau misfolding (manifested as oligomerization) represents, at least partly, a toxic loss of function in the context of synaptic behavior,” he wrote to Alzforum. Pooler suggested that pro-aggregant tau might sequester endogenous mouse tau so that it can no longer perform its normal function. “The data suggest tau plays a role in regulating neuronal signaling, even in the absence of disease,” Pooler said.
However, Decker was quick to point out that the more severe synaptic deficits appeared only in the presence of tau oligomers. “We speculate that the calcium impairment occurs separately from the synaptic transmission phenotype, and explains the impaired plasticity,” he told Alzforum.
In future work, the authors plan to investigate exactly how tau oligomers dampen calcium entry. Eckhard Mandelkow pointed out that as a major microtubule-binding protein, tau participates in the transport of cargo along axons, raising the possibility that some of the observed synaptic changes could be due to a lack of key proteins at the synapse. In support of this, western blots revealed a drop in several synaptic proteins in pro-aggregant mice. The authors will also compare the timing of pre- and postsynaptic deficits to see which one drives the disease.—Madolyn Bowman Rogers
References
News Citations
- Honolulu: The Missing Link? Tau Mediates Aβ Toxicity at Synapse
- The Plot Thickens: The Complicated Relationship of Tau and Aβ
- Tau’s Synaptic Hats: Regulating Activity, Disrupting Communication
- Tau Roundup: Inducible Mice Accentuate Aggregation and More
- Making It Stick—Tau Toxicity Linked to Aggregation Propensity
Research Models Citations
Further Reading
News
- Patient Aβ Dimers Sufficient for Tau, Neuritic Changes
- Grow a Spine—Researchers Explain Brain Boost in Tauopathy Mice
- Aβ and Tau MARK Synapses for Dysfunction
- In Pursuit of Toxic Tau
- Tracing a Path from Aβ to Tau Leads Scientists to Lost Synapses
- Is Dendritic Tau to Blame for AD-Related Hyperexcitability?
- Does Dendritic Tau Promote Plasticity?
- Toxic Tau Quiets Neuronal Networks
Primary Papers
- Decker JM, Krüger L, Sydow A, Zhao S, Frotscher M, Mandelkow E, Mandelkow EM. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol Commun. 2015 Apr 3;3(1):23. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
West Virginia University
This is a very exciting set of experiments describing the role of pathological tau in mediating presynaptic alterations in the DG/CA3 region of the hippocampus. These results add to a recent body of work highlighting the importance of the DG/CA3 and presynaptic activity in cognitive aging and Alzheimer’s disease. At the time point examined in the current study (13 months of age), TauRDΔ mice exhibited a reduction in synaptic vesicles and presynaptic markers and impairments in synaptic transmission, whereas at 2 months of age, no effects of tau were identified. Because some research suggests the DG/CA3 subregion is hyperexcitable in those at risk for Alzheimer’s disease (e.g., Bassett et al., 2006; Bondi et al., 2005; Bookheimer et al., 2000; Filippini et al., 2009; Quiroz et al., 2010; Sperling et al., 2010), it would interesting to investigate activity in the DG/CA3 subregion of TauRDΔ mice to determine whether the DG/CA3 pattern of activity shifts with longer durations of tau expression.
Our work (Hunsberger et al., 2014) suggests that, at least in the rTg(TauP301L)4510 mouse model, early stages of tau pathology are associated with increased glutamatergic activity in the DG/CA3 subregion, and these alterations correlate with behavioral performance in a subregion-specific manner. With longer durations of tau expression and neuron loss, we predict we would see a phenotype closer to that observed by Decker et al. Given the therapeutic implications of increasing or dampening activity at various stages of the disease process, determining how activity patterns of the hippocampus change with disease progression seems an essential next step for this line of research. Similarly, though efforts have been made to understand the DG/CA3 circuit in the disease process, further examination of the recurrent collateral connections of the CA3 will also be important because these collaterals may help to explain the notable sensitivity of the CA3 to tau deposition. As noted by the authors, delineating how these alterations affect behavior, particularly pattern completion and pattern separation, is also critical and may aid in the development of high-throughput screening tests for tracking the progression of hippocampal damage in in human populations.
References:
Bassett SS, Yousem DM, Cristinzio C, Kusevic I, Yassa MA, Caffo BS, Zeger SL. Familial risk for Alzheimer's disease alters fMRI activation patterns. Brain. 2006 May;129(Pt 5):1229-39. PubMed.
Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005 Feb 8;64(3):501-8. PubMed.
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. 2000 Aug 17;343(7):450-6. PubMed.
Filippini N, Macintosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009 Apr 28;106(17):7209-14. PubMed.
Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2015 Jan;132(2):169-82. Epub 2014 Oct 31 PubMed.
Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, Lopera F, Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann Neurol. 2010 Dec;68(6):865-75. PubMed.
Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, Laviolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med. 2010 Mar;12(1):27-43. PubMed.
View all comments by Miranda ReedThe University of Minnesota
The abnormal acetylation of tau lysine 280 (K280) has recently received great attention in AD research. It has been proposed to be a novel pathological signature of AD (see Cohen et al., 2011). The present paper has characterized the effect of deleting tau K280 on long-term depression (LTD) in mossy fiber-CA3 synapses.
The study highlights the role of tau in pre-synaptic plasticity. This is an interesting and new finding, which will help us understand one important aspect of AD and other tauopathies.
Although the authors argue that the changes caused by deltaK280 tau are due to the aggregation of tau proteins, this point needs further substantiation. The acetylation of K280 may activate many unknown signaling pathways, including those that might impair microtubule function and axonal transport, which could be compromised by removing this amino acid.
I would also point out that the plasticity of mossy fiber-CA3 synapses is very different from that in other regions of the brain. Long-term potentiation (LTP) in the majority of excitatory synapses at the cortex and hippocampus is NMDAR-dependent and relies on post-synaptic components. The CA3 region is rare in that LTP is mostly pre-synaptic there and NMDAR-independent. Therefore, the broad significance of K280 deletion in the whole brain remains to be determined. Tau most likely affects both pre-and post-synaptic functions.
References:
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. PubMed.
View all comments by Dezhi LiaoNIMH
It is ironic that despite the overwhelmingly stronger enrichment of tau protein in normal axons, the first evidence for impairments of synaptic functions by hyperphosporylated tau pinpointed dendrites as the problem site. This recent study by Decker et al. now highlights the impact of abnormal tau on presynaptic functions. It presents a series of clear-cut data demonstrating that tau aggregates disrupt the synaptic functions at the presynapses (more so than dendrites), reducing neurotransmitter release and synaptic plasticity at the mossy fiber (MF)-CA3 junction.
Indeed, both basal transmission and activity-dependent changes in MF-CA3 field excitatory postsynaptic potentials (fEPSP) are reduced in transgenic mice expressing aggregation-prone tau in their neurons. The choice of the model is just right: Expression sites of both LTP and LTD at the MF-CA3 connection are presynaptic. Decrease in the paired-pulse ratio of fEPSPs also supports the main conclusion of the paper: The malfunctions have a presynaptic origin (reduced probability of quantal release).
What is quite interesting also about the electrophysiological data of this study is that the threshold for eliciting MF-CA3 field potentials in pro-tau mice (as well as tau-KO mice) is considerably higher (at least 100 percent, see Figure 2a) than non-transgenic controls. This observation delivers an important message—the excitability of neurons (and most likely axons) in transgenic and tau-KO genotypes is lower than in controls, a notion that is in line with recent reports demonstrating that loss of tau can reduce neuronal excitability (i.e., Holth et al., 2013) independently of synaptic functions.
References:
Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013 Jan 23;33(4):1651-9. PubMed.
Make a Comment
To make a comment you must login or register.