No REST for Weary Neurons: Protective Factor Stems Cognitive Decline
Quick Links
Some people seem to possess a “cognitive reserve” that allows them to resist the effects of Alzheimer’s pathology. In the March 20 Nature, researchers led by Bruce Yankner at Harvard Medical School describe a possible molecular mechanism for the phenomenon. The authors found that in aged human brains, the regulatory gene repressor element 1-silencing transcription factor (REST) coordinates a neuroprotective stress response. It turns off genes involved in cell death and pathology and boosts protective factors. High levels of REST correlated with cognitive preservation, even in the presence of amyloid plaques and neurofibrillary tangles. Conversely, people with memory loss had little to no REST. The data hint that the development of dementia requires two hits: Alzheimer’s pathology plus a failure of the brain’s stress-response system, Yankner told Alzforum.
Commentators praised the study’s thoroughness. “This is a broad and in-depth study that goes from patients to animal models, and makes a convincing case for REST downregulation being a general risk factor that makes the aging brain sensitive to neurodegeneration,” Bart de Strooper at VIB and KU Leuven, Belgium, wrote to Alzforum.
REST is not new to science, but it was previously thought to be active only in the developing brain. There, it represses neuronal genes in precursor cells by modifying the surrounding chromatin structure (see Ballas et al., 2005). REST gets degraded as cells differentiate; it is not found in mature neurons. Yankner and colleagues were surprised, therefore, to see REST show up in an aging study. The authors profiled gene expression changes that occur in the prefrontal cortex of aged human brains, and identified a large number of repressed genes (see Lu et al., 2004; Loerch et al., 2008). Many of these contained a motif for REST binding, implying that the transcription factor might regulate a suite of aging-related genes.
In the current paper, first author Tao Lu followed up on these results by examining REST expression in postmortem human brains. In brains from old people, neurons of the prefrontal cortex and hippocampus made five times as much REST as the same neurons in young people, confirming that the factor is induced with age. To find REST’s targets, the authors precipitated DNA bound to the protein. Further analysis revealed that REST shuts down numerous genes involved in cell death, such as FAS, FADD, and cytochrome c. It also silences genes that promote Alzheimer’s pathology, including presenilin 2 and kinases that phosphorylate tau.
Overexpression of REST in neural cell lines pumped up expression of several genes that protect neurons from cell death and oxidative stress, such as BCL2, SOD1, catalase, and FOXO transcription factors. How does REST, a transcriptional repressor, switch on these genes? It may happen indirectly through repression of microRNAs that themselves silence protective genes, since REST is known to act on microRNAs, Yankner told Alzforum. “REST appears to coordinate a network of genes that protect neurons as they age,” Yankner said.
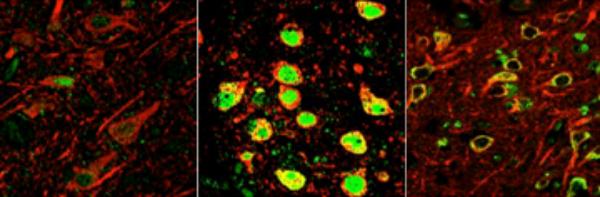
Dormant in young people (left), the transcriptional repressor REST switches on in normal aging human neurons (center) to protect against age-related stresses, including abnormal proteins associated with neurodegenerative disease. In the early stages of Alzheimer's (right), REST is lost in critical brain regions, predisposing to cognitive decline. [Image courtesy of the Yankner Lab.]
The authors then probed REST’s function in several model systems, including primary neuronal cultures, neural cell lines, REST conditional knockout mice, and worms. In all cases, neurons lacking the factor died more quickly when exposed to stressors such as hydrogen peroxide or oligomeric Aβ42. Knockout mice developed neurodegeneration around eight months of age, and worms missing the REST ortholog SPR-4 expired twice as fast as control animals when exposed to oxidative stress. In all these models, REST expression rescued neurons. Oxidative stress, which increases with age, has been implicated as an AD risk factor (see, e.g., Apr 2012 news story).
Given REST’s apparent broad protective function, what specific role, if any, does the protein play in neurodegenerative disease? In several dozen AD brains tested postmortem, the authors saw that REST protein was absent from the nuclei of cortical and hippocampal neurons. They did spot the protein, clumped with misfolded Aβ, in autophagosomes, the cellular degradation organelles of the lysosomal/autophagy pathway. Since autophagy is implicated in several neurodegenerative diseases, the authors also examined brain slices from people with frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB). In each case, they saw REST in autophagosomes along with the characteristic pathologic protein of that disease. Together, the data suggest to Yankner that the presence of pathogenic proteins might somehow consign REST for disposal by autophagy.
How early in disease might this happen? One clue comes from the brains of people who died with amnestic mild cognitive impairment (MCI). In cortical and hippocampal neurons, nuclear REST was down about 50 percent compared with healthy aged brains.
The authors next correlated REST expression with cognition using data from the Religious Orders Study and the Rush Memory and Aging Project. In these longitudinal studies, volunteers undergo serial cognitive testing and donate their brains after death. Participants who died with more REST in the nuclei of their cortical and hippocampal neurons turned out to have done better on cognitive tests, particularly in episodic memory. They were also less likely to have AD pathology. Notably, participants who had plaques and tangles but little cognitive impairment had three times more nuclear REST than did those with dementia, supporting the idea that REST preserves cognition in the face of AD pathology for some time.
Finally, Yankner and colleagues searched for proteins that might control REST. They found that stressed cells released soluble factors that induced this transcription factor in unexposed neurons. REST induction could be stimulated by purified Wnt-3a and Wnt-7a and blocked by inhibitors of Wnt signaling. This fits with other work suggesting that REST is a target of Wnt signaling (see Willert et al., 2002). Some drugs activate this signaling pathway, for example, the mood stabilizer lithium. Yankner noted that some studies have found a lower incidence of AD in people on lithium (see Terao et al., 2006; Yeh and Tsai, 2008; Nunes et al., 2013). Yankner is conducting a high-throughput screen to look for other molecules that boost REST expression and might have therapeutic potential.
In an accompanying editorial, Li-Huei Tsai and Ram Madabhushi at Massachusetts Institute of Technology, Cambridge, note that too much Wnt signaling can trigger cancer, so therapeutic approaches would have to be carefully calibrated. “A deeper understanding of the molecular mechanisms that govern REST activation in the aging brain will be crucial for such efforts to be successful,” they wrote.—Madolyn Bowman Rogers.
References
News Citations
Paper Citations
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005 May 20;121(4):645-57. PubMed.
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004 Jun 24;429(6994):883-91. PubMed.
- Loerch PM, Lu T, Dakin KA, Vann JM, Isaacs A, Geula C, Wang J, Pan Y, Gabuzda DH, Li C, Prolla TA, Yankner BA. Evolution of the aging brain transcriptome and synaptic regulation. PLoS One. 2008;3(10):e3329. PubMed.
- Willert J, Epping M, Pollack JR, Brown PO, Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol. 2002 Jul 2;2:8. PubMed.
- Terao T, Nakano H, Inoue Y, Okamoto T, Nakamura J, Iwata N. Lithium and dementia: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry. 2006 Aug 30;30(6):1125-8. Epub 2006 Jun 6 PubMed.
- Yeh HL, Tsai SJ. Lithium may be useful in the prevention of Alzheimer's disease in individuals at risk of presenile familial Alzheimer's disease. Med Hypotheses. 2008 Dec;71(6):948-51. PubMed.
- Nunes MA, Viel TA, Buck HS. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer�s disease. Curr Alzheimer Res. 2012 Jun 29; PubMed.
Further Reading
News
- Multitalent RNA: Guides Stem Cells, Cuts Itself to Turn Off Gene
- Dementia and Education—What You Don’t Know May Speak Volumes
- Social Seniors: Strong Networks Build Cognitive Reserve
- Brain-Specific Sirtuin Expression Delays Aging in Mice
- Histone-Binding Protein Slows 'Normal Aging'?
- Could Longevity Factor, Epilepsy Med, Treat AD One Day?
- A Boost for Waning DNA Tags Fixes Age-Related Memory Loss in Mice
- Neurodegeneration and Aging: Could MicroRNA Be the Link?
Primary Papers
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiácovo MP, Yankner BA. REST and stress resistance in ageing and Alzheimer's disease. Nature. 2014 Mar 27;507(7493):448-54. Epub 2014 Mar 19 PubMed.
- Tsai LH, Madabhushi R. Alzheimer's disease: A protective factor for the ageing brain. Nature. 2014 Mar 27;507(7493):439-40. Epub 2014 Mar 19 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Roche
A sizable proportion of elderly individuals with substantial AD pathology does not appear to progress to dementia, indicating that specific neuroprotective mechanisms must exist to evade progressive cognitive decline. The elegant and comprehensive study by Lu et al. provides convincing evidence that the developmental transcriptional factor REST, together with epigenetic repression of REST target genes, regulates a neuroprotective stress response that appears to be essential for proper neuronal functions and cognitive preservation during aging. The authors demonstrate that REST is induced in specific neuronal populations of the aging human brain to confer oxidative stress resistance and protection against toxic insults associated with aging and AD. Intriguingly, in individuals who met pathological criteria for AD, nuclear REST was significantly elevated in cognitively preserved, as compared to cognitively impaired, subjects, suggesting that failure of the brain’s cellular stress response system underlies the transition from successful to pathological brain aging.
These exciting findings provide a molecular signature of a crucial stress-response program that will certainly advance the field toward the identification and further characterization of neuroprotective signaling pathways with highest therapeutic relevance for AD and other aging-associated neurodegenerative diseases. Besides oxidative stressors, the authors identified Wnt-β-catenin signaling as an inducer of REST in the aging brain. It will be highly relevant to see whether other neurodevelopmental programs that are maintained in the adult brain to sustain synaptic plasticity and cognitive functions are also involved in maintaining REST-mediated signaling in the aging brain.
Yale University School of Medicine
There are a number of puzzling aspects of the manuscript from the Yankner lab and the comments provided by different reviewers. The most remarkable is the lack of any citation, either by the authors of the manuscript, or any of the commentators, of earlier studies that show REST to be activated in adult neurons by ischemia (Calderone et al., 2003; Noh et al., 2012). How come? If these findings are thought to be suspect, the authors should explain why. If not they probably explain why aging brains express REST. In any case it is premature to label REST a “protective factor”, so far it is at best correlative. There are many uncertainties that remain to be explored.
References:
Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003 Mar 15;23(6):2112-21. PubMed.
Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012 Apr 17;109(16):E962-71. Epub 2012 Feb 27 PubMed.
Make a Comment
To make a comment you must login or register.