New WAVE in Aβ Production—AICD Tempers APP Processing
Quick Links
When the amyloid precursor protein (APP) gets chopped up to make Aβ, other fragments are released, including APP’s intracellular domain. Scientists believe AICD acts as a transcription factor, though its genomic targets are poorly characterized. In the August 17 Nature Medicine, researchers led by Yong Kim and Paul Greengard at Rockefeller University in New York report that AICD regulates the gene for WAVE1, a protein needed to transport APP from the Golgi to the plasma membrane. AICD suppresses expression of WAVE1, reducing APP at the membrane and thus its processing, providing negative feedback for Aβ production, according to the researchers. Other scientists found the transcriptional regulation intriguing, but stressed the need for more supporting evidence that it modulates Aβ production.
“This reinforces the view that AICD is an important transcriptional regulator and a key component of the actions of APP,” wrote Anthony Turner, University of Leeds, England, who was not involved in the study. While it is still unclear whether the overall effects of AICD are neuroprotective or neurodegenerative, this study is an important step forward, he said.
WAVE1, short for the mouthful Wiskott-Aldrich syndrome protein (WASP)-family verprolin homologous protein 1, activates the Arp2/3 complex, which initiates polymerization of actin. Among other things, this polymerization helps vesicles bud from the Golgi and carry proteins to the plasma membrane. In the brain, WAVE1 exists in a complex with four other proteins. One of them, NCKAP1, was previously reported to be downregulated in the brains of people with Alzheimer’s disease (Suzuki et al., 2000). Kim’s group wondered whether WAVE1 expression also fell in AD, and what the consequences might be.
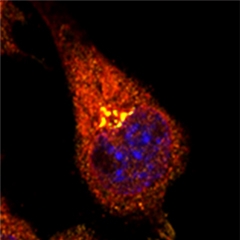
Bound in the Golgi.
In neuroblastoma cells, WAVE1 (orange) and APP (red) co-localize in the Golgi. The nucleus is labeled in blue. [Courtesy of Ceglia et al., Nature Medicine.]
In 3xTg and Tg2576 AD mouse models, which overexpress the Swedish mutant of APP, first author Ilaria Ceglia and colleagues found that WAVE1 levels fell by 25 percent to 30 percent relative to wild-type mice. In postmortem human brain tissue, WAVE1 levels were also lower than in tissue taken from healthy controls, but only in regions affected by AD. Likewise, neuroblastoma cells (N2a) expressing wild-type APP contained half the WAVE1 of non-transfected cells. When Ceglia treated these cells with a γ-secretase inhibitor, WAVE1 levels returned to normal. Together, these data hint that APP somehow shuts down WAVE1 expression.
What explains these results? The group suspected that APP’s intracellular domain had something to do with it. AICD reportedly up- or downregulates a variety of genes, such as the Aβ-degrading enzyme neprilysin and the tumor suppressor p53 (for a review see Pardossi-Piquard and Checler, 2012). Using chromosome immunoprecipitation assays, Ceglia found that AICD interacted with the WAVE1 gene promoter. When she overexpressed AICD it brought WAVE1 crashing down.
Ceglia and colleagues next wondered how this drop in WAVE1 affected the cell. In neuroblastoma cells overexpressing APPSwe and PS1ΔE9, tuning down WAVE1 with small interfering RNAs reduced the amount of Aβ40, Aβ42, and sAPPβ they churned out. This implied less β-secretase cleavage. Likewise, APP/PS1 mice missing one copy of WAVE1 did not produce as much Aβ40 and Aβ42 as control transgenic animals, and mice missing both WAVE1 genes produced even less. “Because we didn’t see a significant change in the soluble APP generated by α-secretase, we think WAVE1 downregulation affects the amyloidogenic pathway,” said Kim.
Immunoprecipitation experiments revealed that WAVE1 and APP bound each other in the Golgi (see image above). In in vitro assays, knocking down WAVE1 reduced APP-containing vesicles budding from the Golgi membrane. Since trafficking of APP from the Golgi to the plasma membrane and endosomes exposes the precursor to secretases, limiting that transport could reduce formation of Aβ, Kim told Alzforum (see image below). In fact, genetically halving WAVE1 corrected memory deficits in the Morris water maze in the APP/PS1 double transgenics.
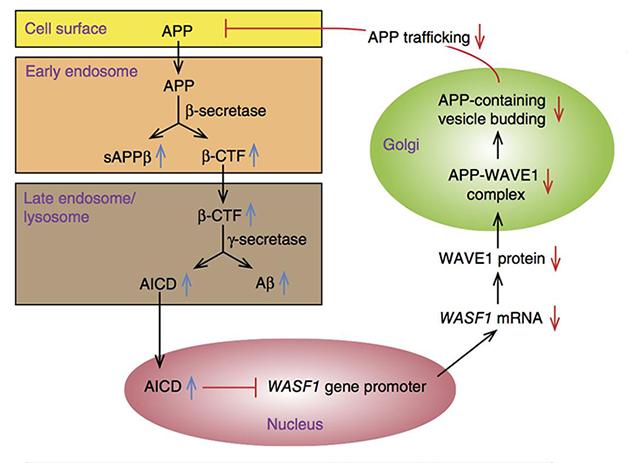
Feedback. APP processing generates AICD, which curbs expression of WAVE1, reduces APP trafficking from the Golgi, and reduces Aβ production. [Courtesy of Ceglia et al., Nature Medicine.]
Kim suggested this protective feedback may be altered or overpowered in AD, an idea he plans to explore in future experiments. The pathway may suggest a way to therapeutically reduce formation of Aβ, perhaps by interfering with the WAVE1-APP interaction to inhibit trafficking, the authors wrote.
“This study supports previous data that, like the Notch intracellular domain and other γ-secretase-derived intracellular domains, AICD can act as a transcriptional regulator," wrote Wim Annaert, KU Leuven, Belgium. He cautioned that the work relied on overexpression models and wondered whether enough AICD—which has a short half-life—reaches the nucleus under physiological conditions to affect gene transcription. He also noted that WAVE1 complexes affect trafficking at other sites in the cell, meaning they could, for instance, promote APP internalization at the plasma membrane (see full comment below). Monitoring APP surface levels and dynamics of internalization would help flesh out the picture.
Ralph Nixon, New York University School of Medicine, suggested that the feedback mechanism may not exist to modulate Aβ production. More likely it exists to modulate APP or another of its metabolites important for signaling, such as AICD itself or the β-C-terminal fragment, he said. He thought it would be interesting to know how these other metabolites are affected by reducing WAVE1 and how the potential changes influence neuronal function.—Gwyneth Dickey Zakaib
References
Research Models Citations
Paper Citations
- Suzuki T, Nishiyama K, Yamamoto A, Inazawa J, Iwaki T, Yamada T, Kanazawa I, Sakaki Y. Molecular cloning of a novel apoptosis-related gene, human Nap1 (NCKAP1), and its possible relation to Alzheimer disease. Genomics. 2000 Jan 15;63(2):246-54. PubMed.
- Pardossi-Piquard R, Checler F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012 Jan;120 Suppl 1:109-24. PubMed.
Further Reading
Papers
- Buoso E, Lanni C, Schettini G, Govoni S, Racchi M. beta-Amyloid precursor protein metabolism: focus on the functions and degradation of its intracellular domain. Pharmacol Res. 2010 Oct;62(4):308-17. PubMed.
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18367-72. PubMed.
- Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci. 2009 Oct 15;122(Pt 20):3703-14. PubMed.
- Li M, Pehar M, Liu Y, Bhattacharyya A, Zhang SC, O'Riordan KJ, Burger C, D'Adamio L, Puglielli L. The amyloid precursor protein (APP) intracellular domain regulates translation of p44, a short isoform of p53, through an IRES-dependent mechanism. Neurobiol Aging. 2015 Oct;36(10):2725-36. Epub 2015 Jun 21 PubMed.
- Grimm MO, Mett J, Stahlmann CP, Grösgen S, Haupenthal VJ, Blümel T, Hundsdörfer B, Zimmer VC, Mylonas NT, Tanila H, Müller U, Grimm HS, Hartmann T. APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front Aging Neurosci. 2015;7:77. Epub 2015 May 19 PubMed.
- Shu R, Wong W, Ma QH, Yang ZZ, Zhu H, Liu FJ, Wang P, Ma J, Yan S, Polo JM, Bernard CC, Stanton LW, Dawe GS, Xiao ZC. APP intracellular domain acts as a transcriptional regulator of miR-663 suppressing neuronal differentiation. Cell Death Dis. 2015 Feb 19;6:e1651. PubMed.
Primary Papers
- Ceglia I, Reitz C, Gresack J, Ahn JH, Bustos V, Bleck M, Zhang X, Martin G, Simon SM, Nairn AC, Greengard P, Kim Y. APP intracellular domain-WAVE1 pathway reduces amyloid-β production. Nat Med. 2015 Sep;21(9):1054-9. Epub 2015 Aug 17 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Institut de Pharmacologie Moléculaire et Cellulaire
The article by Ceglia and colleagues is an informative and interesting work from several vantagepoints. First is the crystal-clear demonstration that AICD controls WAVE1 expression at a transcriptional level. This adds to the increasing list of well- characterized AICD transcriptional targets (for review see Pardossi-Piquard et al., 2012) and highlights the need for more epigenetic studies aimed at delineating the molecular determinants/co-factors underlying AICD activator and repressor transcriptional functions.
It is notable that we previously reported that AICD enhances the transcription of the Aβ-degrading enzyme neprilysin (Pardossi-Piquard et al. 2005). Thus, at a first glance AICD appears to lower Aβ levels at various steps by favoring NEP-mediated Aβ catabolism and by lowering WAVE-1-linked Aβ production.
However, this work does not definitely establish whether AICD is beneficial or deleterious. Overall, the data could indicate, as suggested by the authors, that AICD contributes to a feedback process aimed at thwarting Aβ load. Conversely, this view is apparently challenged by both conceptual and experimental arguments suggesting a pathogenic role for AICD. First, in sporadic AD, it is generally believed that increased Aβ load results from alterations of post-production processes such as lowered catabolism rather than from increased production. Second, AICD hippocampal levels are likely increased in AD brains due to age-dependent lowering of its catabolizing enzyme, insulin-degrading enzyme (Cook et al, 2003). Third, we and others showed that AICD mainly derives from the amyloidogenic pathway (Goodger et al., 2009; Belyaev et al., 2010; Flammang et al., 2012). Fourth, familial cases of AD linked to mutations in APP or presenilins usually increase γ-secretase-mediated production of Aβ and its counterpart AICD. Fifth, even AICD-mediated enhancement of NEP activity, which could be seen as a beneficial process, results in an increased Aβ42/Aβ40 ratio (because Aβ40 is more readily cleaved than Aβ42), a cellular parameter driving pathology at least in cells (Kuperstein et al., 2010). Sixth, AICD overexpression recapitulates most AD-related stigmata in transgenic mice (Ghosal et al., 2009). Overall, the data strongly argue in favor of a pathogenic contribution of AICD, at least at late stages of the disease.
A possibility remains that AICD-mediated control of WAVE-1 could be part of a compensatory mechanism aimed at reducing Aβ load, at least at early stages of the disease. However, one cannot exclude the possibility that the genuine lowering of the pool of membranous APP triggered by a WAVE-1-dependent, AICD-mediated process could also reflect an enhanced rate of processing occurring in other cell compartments yielding additional catabolites such as C99 that is likely contributing early to AD pathology (Lauritzen et al. 2012).
In conclusion, the observation by Ceglia and colleagues is extremely interesting and opens a new potential track in the amyloidogenic hypothesis, but additional studies are clearly necessary before considering AICD/WAVE-1/APP cascade as a novel therapeutic target.
References:
Pardossi-Piquard R, Checler F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012 Jan;120 Suppl 1:109-24. PubMed.
Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves DA Costa C, Vincent B, Ring S, D'Adamio L, Shen J, Müller U, St George Hyslop P, Checler F. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005 May 19;46(4):541-54. PubMed.
Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, Schellenberg GD, Jin LW, Kovacina KS, Craft S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003 Jan;162(1):313-9. PubMed.
Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci. 2009 Oct 15;122(Pt 20):3703-14. PubMed.
Belyaev ND, Kellett KA, Beckett C, Makova NZ, Revett TJ, Nalivaeva NN, Hooper NM, Turner AJ. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a {beta}-secretase-dependent pathway. J Biol Chem. 2010 Dec 31;285(53):41443-54. PubMed.
Flammang B, Pardossi-Piquard R, Sevalle J, Debayle D, Dabert-Gay AS, Thévenet A, Lauritzen I, Checler F. Evidence that the Amyloid-β Protein Precursor Intracellular Domain, AICD, Derives From β-Secretase-Generated C-Terminal Fragment. J Alzheimers Dis. 2012 Jan 1;30(1):145-53. PubMed.
Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010 Oct 6;29(19):3408-20. PubMed.
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18367-72. PubMed.
Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, Chami L, Dunys J, Checler F. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012 Nov 14;32(46):16243-55a. PubMed.
KULeuven & VIB
Overall, and like several others in recent years, this new study highlights the importance of intracellular transport regulation in the regulation of APP processing. Transport (dys)regulation is clearly an emerging theme not only in AD but also in other neurodegenerative diseases like PD, ALS, FTD, and others. This is, in the end, not surprising since proper transport ensures cellular homeostasis and provides multiple ways for the neuron to get rid of dysfunctional organelles and/or misfolded/aggregated proteins and peptides. Failure to do so initiates an inevitable and irreversible route to cell death and degeneration. This is also evident from GWAS studies, where the products of novel susceptibility genes often relate to (endosomal) transport regulation. These unbiased approaches do not always mirror candidate approaches (like here with WAVE1), but that is not necessarily problematic: The future challenge will be to determine how these susceptibility genes cross paths with other transport regulations or signaling cascades.
The current study demonstrates for the first time a negative feedback loop for Aβ production through AICD-mediated transcriptional regulation of WAVE1 expression. It also supports previous data that, like Notch intracellular domain and other γ-secretase-derived intracellular domains, AICD also acts as a transcriptional regulator. Some caution is still needed since this study relies entirely on (high) overexpression models. Given the very short half-life of AICD, it remains to be seen whether under physiological conditions sufficient AICD is available to translocate to the nucleus and regulate transcription. The study limits itself to studying WAVE1 expression, though WAVE1 functions in a heteropentameric complex: How the other components of this complex behave, and to what extent the level of functional complexes is affected by AICD, are some upcoming intriguing questions.
The authors have analyzed sporadic AD brain but an extension of the study to PSEN-FAD is an obvious next step. Many FAD-PSEN1 mutations do not result in more Aβ production, but rather shift the balance to more neurotoxic species due to, for instance, a drop in Aβ40 production. It follows that within the amyloidogenic pathway, AICD’s of different lengths are produced: A question that pops up is whether these different forms equally affect WAVE1 expression or not.
In their model, the authors conclude that a drop in WAVE1 during disease progression may provide negative feedback, suggesting that it may slow down disease. It would be interesting to find out at what point, or BRAAK stage, WAVE1 levels start to drop, because moving forward that deflection point could have therapeutic value. But one cannot be too over-enthusiastic here, since WAVE1 complexes play critical roles in neuronal development, including migration and dendritic spine morphology, which one doesn’t want to compromise too much.
The authors provide evidence that the drop in WAVE1 levels upon overexpression of APP affects Golgi exit and the biogenesis of AP1-dependent Golgi carriers. This is a valid conclusion, but in my opinion incomplete because it is not supported by complementary data monitoring APP surface levels and dynamics of internalization. In this respect, it is important to appreciate that WAVE1 complexes act at the cell surface as well, where they could modulate internalization as part of cellular migration. Alternatively, WAVE1 may integrate several signaling pathways affecting APP trafficking and processing at different locations. In this regard, it is interesting to consider the study of Deyts et al., who showed a non-transcriptional role for the membrane-tethered APP-CTF (and thus the precursor of AICD) through the coupling of G-protein to adenylate cyclase and cAMP signaling that results in protein kinase A activation and inhibition of GSK3beta signaling, ultimately affecting dendritic arborization (Deyts et al., 2012). Since cAMP signaling reduces CDK5 phosphorylation of WAVE1 in another study, APP expression and processing could regulate WAVE1 function in different ways.
Finally, the effect of WAVE1 downregulation on Golgi exit should be studied in the light of polarized neuronal trafficking. It is well established by work of the Bonifacino group at the NIH, Bethesda, Maryland, that the Golgi-associated AP1 complex directs cargo to dendrites: Abrogating this interaction diverts cargo to axons as well. The same group also demonstrated an interaction of APP with the AP4 complex—thus, and specifically for neurons, an alternative (or supplementary) explanation for the effect of WAVE1 downregulation on APP processing could be altered delivery of APP, thereby affecting its encounter with amyloidogenic secretases.
References:
Deyts C, Vetrivel KS, Das S, Shepherd YM, Dupré DJ, Thinakaran G, Parent AT. Novel GαS-protein signaling associated with membrane-tethered amyloid precursor protein intracellular domain. J Neurosci. 2012 Feb 1;32(5):1714-29. PubMed.
Case Western Reserve University
I believe the statement “… still unclear whether the overall effects of AICD are neuroprotective or neurodegenerative …” is incorrect. In a series of papers starting with Ghosal et al., 2009, we have shown that AICD transgenic mice recapitulate major AD-pathological features, including memory deficits and neurodegeneration, in an age-dependent manner (Vogt et al., 2011; Ghosal et al., 2011; Ghosal et al., 2010; Margevicius et al., 2015). Several other groups have independently shown neurotoxic effects of AICD in vitro in neuronal and non-neuronal cells. The overall effects of AICD are demonstrably harmful.
This is an intriguing study linking AICD to reduced Aβ generation via the WAVE1 pathway. However, a crucial piece of data is missing—that AICD reduces Aβ levels in vivo in the brains of mice. By crossing AICD-Tg mice with APP/PS1 and R1.40 mice models of AD, we found no reduction in Aβ40, Aβ42, or plaque load (Figure 6 in Ghosal et al., 2009). In fact, there are no data in the present study directly linking AICD expression with reduced Aβ: Ceglia et al. only show reduced WAVE1 protein in N2a cells transfected with AICD (Fig. 1g). Based only on this piece of data, it is premature to conclude that AICD reduces Aβ production in vivo.
The authors do present a very interesting piece of data buried inside the figures. By knocking down one copy of the WAVE1 gene, Wasf1, in APPswe/PS1dE9 2xTg mice, they observe greater reduction of Aβ40 levels compared to that of Aβ42 levels (Fig. 2h versus 2i). This results in an increased ratio of Aβ42/40 (from approximately 8 in 2xTg mice to 13 in 2xTg;Wasf1 +/-), which is considered harmful and a potential cause of AD in families with FAD mutations. Surprisingly, the deletion of one copy of Wasf1 rescued the behavioral deficits in Morris Water maze in the 2xTg mice (Fig. 4a, 4b). Does it mean that increased Aβ42/40 ratio is protective for mice? This finding begs for a better explanation.
References:
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18367-72. PubMed.
Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011 Sep;32(9):1725-9. PubMed.
Ghosal K, Pimplikar SW. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol Aging. 2011 Dec;32(12):2320.e1-9. PubMed.
Ghosal K, Stathopoulos A, Pimplikar SW. APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One. 2010;5(7):e11866. PubMed.
Margevicius DR, Bastian C, Fan Q, Davis RJ, Pimplikar SW. JNK-interacting protein 1 mediates Alzheimer's-like pathological features in AICD-transgenic mice. Neurobiol Aging. 2015 Aug;36(8):2370-9. Epub 2015 Apr 30 PubMed.
Make a Comment
To make a comment you must login or register.