From New Tau Structure, Bona Fide Aggregation Inhibitors?
Quick Links
In another win for cryo-electron microscopy, researchers led by David Eisenberg at the University of California, Los Angeles, have solved the structure of a 10-residue tau segment containing the famed VQIINK hexapeptide, which lies at the beginning of tau’s second microtubule-binding domain. As reported in the November 20 Nature Chemistry, a steric zipper with strong intermeshed teeth may explain how tau molecules form fibrils. Three different zippers in the region emerged from the structural analysis, and the researchers designed peptide inhibitors to disrupt them. Strikingly, they prevented the formation of fibrils of full-length tau both in vitro and in cells.
- A cryo-EM structure of fibrils formed from a 10-residue segment of tau suggests the VQIINK hexapeptide forms tight steric zippers.
- The zipper region promoted fibril formation in vitro.
- Zipper inhibitors prevented propagation of tau fibrils in biosensor cells.
Though the findings place the VQIINK peptide at the heart of tau aggregation, some consider them at odds with a recently reported cryo-EM structure of tau fibrils from an AD patient (Jul 2017 news) that placed the sequence outside of the fibril’s core. Nonetheless, researchers thought Eisenberg’s decapeptide structures may be important. “It is great work,” noted Marc Diamond, University of Texas Southwestern Medical Center in Dallas. “While I’m not sure about the overall significance for the structure of tau in tauopathies, since their experiments (by necessity) were done with very simplified tau structural elements, the efficacy of the inhibitors suggests their structural predictions have validity,” he said.
Tau aggregation underlies tauopathies, and efforts to develop drugs that inhibit this process abound. Eisenberg and others have focused on the structures of key aggregation-promoting regions of tau (Sep 2012 news). However, specific isoforms of the protein predominate in different tauopathies. In its full glory, tau contains four microtubule-binding domains, but alternative splicing also creates a shorter, three-repeat (3R) tau that lacks the second of these domains. Each of the four repeats begins with a slightly different hexapeptide sequence. Two of these, VQIINK on the second repeat (R2), and VQIVYK on R3, are known to promote tau aggregation (von Bergen et al., 2000; von Bergen et al., 2001). A decade has passed since Eisenberg’s group used X-ray micro-diffraction to solve the structure of VQIVYK, and subsequently develop aggregation inhibitors (Sawaya et al., 2007; Jun 2011 news). These blocked the aggregation of 3R but not 4R tau, which contains the VQIINK on R2.
VQIINK has resisted efforts to crystalize it. Now, first author Paul Seidler and colleagues have capitalized on micro-electron diffraction, a form of cryo-electron microscopy that makes do with the tiniest of crystals, to solve the structure of nanocrystals of a 10-residue KVQIINKKLD peptide to 1.5 Ångstroms. As with R3’s VQIVYK hexapeptide, R2’s VQIINK formed a Type I, homosteric zipper, in which two of the peptides aligned anti-parallel to each other, with their protruding side chains forming the teeth of the zipper. This zipper turned out to be heavy-duty compared with VQIVYK’s: its VQIIN side chains interwove more tightly and a greater proportion of them slid together. The researchers dubbed this zipper “interface A.”
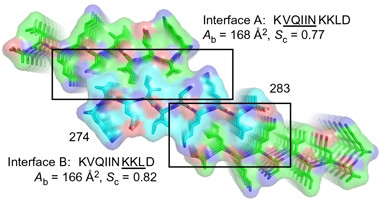
Double zippers.
By virtue of two steric zippers, denoted by interface A (top) and B (bottom), any tau molecule (turquoise) can bind two others (green). The highly stable structure may then form fibrils (denoted here by stacking). [Courtesy of Seidler et al., Nature Chemistry, 2017.]
Surprisingly to Eisenberg, the researchers found that a second zipper, “interface B,” formed at the C-terminal end of the motif. As for interface A, but this time utilizing residues KKLD, the interface comprised two peptides zipped together in anti-parallel fashion. Because the two interfaces projected from opposite side of the peptide, both zippers can close at the same time, effectively stitching any tau molecule simultaneously to two others (see image at right). Eisenberg was uncertain what this double-zipper arrangement would portend for aggregation, or how common the resulting peptide troika would be. However, in vitro fibrillization experiments confirmed that VQIINK promoted fibril formation more robustly than VQIVYK did. Models predicted that R3’s VQIVYK hexapeptide cannot form this second zipper.
Computational modeling also indicated that deleting lysine280, which lies at the junction between the two zippers, would flip interface B to the same side of the peptide as interface A, creating one long zipper. K280 deletion causes frontotemporal dementia and kicks tau aggregation into overdrive (Barghorn et al., 2000). Eisenberg thinks this megazipper could explain why tauDK280 fibrillizes so readily.
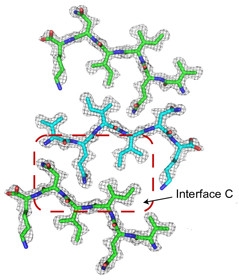
And then there were three …
Nanocrystals of VQIINK revealed a third zipper, interface C, formed in a face-to-back configuration. [Courtesy of Seidler et al., Nature Chemistry, 2017.]
Subsequently, the researchers detected a second structure of the peptide from a separate batch of nanocrystals, this time using only the six amino acids, VQIINK. In this they identified yet a third zipper. Employing side chains from QII residues as teeth, “interface C” formed in a “face-to-back” configuration, mating the interface A side of one peptide to the interface B side of another (see image at right). Eisenberg plans to investigate whether the different zipper interfaces define strains of tau found among people with different tauopathies or even in people with specific tauopathies.
The recently reported structure of tau fibrils from an AD patient placed the R2 domain (which contains VQIINK) out in a structurally heterogeneous “fuzzy coat,” rather than the fibril’s core. How could VQIINK play a pivotal role in fibrillization from there? Eisenberg speculated that perhaps VQIINK seeds aggregation of new fibrils from its more exposed locale, without incorporating into the fibril’s core. Either that, or the two structures represent different structural strains of tau, he suggested. Diamond was not so sure about the former interpretation, noting it is difficult to reconcile that VQIINK plays a role in initiating aggregation, but is not required to form the core of filaments.
Unzip It
Can these zippers be undone? The researchers used the cryo-EM structure to design peptide inhibitors to bind VQIINK and disrupt the anti-parallel interfaces. They initially focused on interfaces A and B, designing zipper-disrupting peptides with sequences WINK and MINK. MINK slightly outperformed WINK, halving aggregation of full-length tau in in vitro assays. In biosensor cell lines, developed by Diamond when he was at Washington University, St. Louis (Oct 2014 news), 125μM MINK more than halved aggregation of tau in the cytoplasm. Notably, the inhibitor even blocked the seeding activity of preformed fibrils, considered the most potent seeds, though more than 20μM MINK was required to achieve half maximal inhibition. Ultimately, the researchers improved efficacy by about 20-fold by re-engineering the inhibitor to block interface C as well. At 2.5μM, the peptide W-MINK completely blocked tau aggregation induced by preformed tau fibrils (see image below). The findings suggest that thoroughness in structural studies pays off, Eisenberg said, because targeting all three zippers produced the most potent inhibitor.

Aggregation Inhibitor. Fibrils of full-length tau seeded aggregation in biosensor cell lines in presence of 0.3μM W-MINK (left), but not 2.5μM (right). [Courtesy of Seidler et al., Nature Chemistry 2017.]
Eisenberg thinks the VQIINK-targeted inhibitors, especially because they target multiple zipper interfaces, might be effective against diverse structural polymorphs, or strains, of tau. ADRx, Inc., a start-up Eisenberg co-founded, is developing such inhibitors.
Eisenberg previously developed VQIVYK-targeted inhibitors, but these failed to block fibrillization of full-length tau. However, it is still unclear which hexapeptide makes the best target, according to Aileen Funke of Coburg University in Germany. Funke was impressed by the elucidation of the VQIINK structure and development of the corresponding inhibitors, but added that her lab has derived a VQIVYK-based inhibitor that does block seeding of full-length tau, at least in vitro (Dammers et al., 2016).—Jessica Shugart
References
News Citations
- Tau Filaments from the Alzheimer’s Brain Revealed at Atomic Resolution
- Research Brief: “Hao” Do You Like Them Amyloid Blockers?
- Structure-Based Approach Yields Tau Inhibitors
- Cellular Biosensor Detects Tau Seeds Long Before They Sprout Pathology
Mutations Citations
Paper Citations
- von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A. 2000 May 9;97(10):5129-34. PubMed.
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem. 2001 Dec 21;276(51):48165-74. PubMed.
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AØ, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007 May 24;447(7143):453-7. PubMed.
- Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000 Sep 26;39(38):11714-21. PubMed.
- Dammers C, Yolcu D, Kukuk L, Willbold D, Pickhardt M, Mandelkow E, Horn AH, Sticht H, Malhis MN, Will N, Schuster J, Funke SA. Selection and Characterization of Tau Binding ᴅ-Enantiomeric Peptides with Potential for Therapy of Alzheimer Disease. PLoS One. 2016;11(12):e0167432. Epub 2016 Dec 22 PubMed.
Further Reading
Primary Papers
- Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, Gonen T, Eisenberg DS. Structure-based inhibitors of tau aggregation. Nat Chem. 2018 Feb;10(2):170-176. Epub 2017 Nov 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Institut für Strukturbiologie und Biophysik
Aggregated tau protein plays a significant role in a variety of neurological disorders, including Alzheimer’s disease. The development of specific compounds to inhibit tau aggregation can aid the development of therapies and might help to elucidate the pathologic mechanisms of the various tauopathies. The sequence segments VQIVYK and VQIINK are well known to drive tau aggregation, and peptides were already developed to block tau aggregation by binding to the VQIVYK segment, which is central to the core of tau fibrils extracted from AD brain tissue (Fitzpatrick et al., 2017; Sievers et al., 2011; Dammers et al., 2016).
In this recent article, Seidler et al. described the structure of VQIINK containing fibrils, resolved by micro-electron diffraction (microED). The segment formed a face-to-face Type 1 homosteric zipper, like VQIVYK (Sievers et al., 2011), but with a stronger interface. The determination of the VQIINK fibril structure is a great success and the result of much hard work (for a decade, efforts of the group were unsuccessful), and was made possible only by the invention of microED. The authors used the structure to design peptide inhibitors of tau aggregation, which inhibited aggregation of full-length tau but also the ability of exogenous full-length tau fibrils to seed intracellular tau in HEK293 biosensor cells into amyloid fibers. These results were compared to the performance of a VQIVYK binding peptide D-TLKIVW, which was also designed to block tau aggregation (Sievers et al., 2011). The latter peptide was not able to block full-length tau aggregation, nor could it inhibit tau seeding in the HEK293 biosensor model.
In addition, the authors developed tau K18 isoforms that possessed two VQIINK or two VQIVYK motifs and found that the variant with two VQIINK motifs fibrillized more rapidly, whereas the other variant formed more tau oligomers (the more toxic species?). The authors concluded that the VQIINK fragment was the more powerful driver of tau aggregation and a superior target for the development of tau aggregation inhibitors.
Which of the motifs is the superior target for tau aggregation inhibitor development is to be proven in the future. Recently, we have developed D-enantiomeric peptides, which were selected to bind to VQIVYK fibrils. These peptides, in contrast to D-TLKIVW, were able to inhibit full-length tau fibrillization (Dammers et al., 2016). Their potential to inhibit the ability of exogenous full-length tau fibrils to seed intracellular tau is not tested yet, but might interesting to evaluate.
References:
Fitzpatrick AW, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SH. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. 2017 Jul 13;547(7662):185-190. Epub 2017 Jul 5 PubMed.
Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Münch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011 Jul 7;475(7354):96-100. PubMed.
Dammers C, Yolcu D, Kukuk L, Willbold D, Pickhardt M, Mandelkow E, Horn AH, Sticht H, Malhis MN, Will N, Schuster J, Funke SA. Selection and Characterization of Tau Binding ᴅ-Enantiomeric Peptides with Potential for Therapy of Alzheimer Disease. PLoS One. 2016;11(12):e0167432. Epub 2016 Dec 22 PubMed.
Make a Comment
To make a comment you must login or register.