LRRK2 Interactions Identify New Parkinson’s Genes, Implicate Autophagy
Quick Links
Although the most common genetic cause of familial Parkinson’s, LRRK2 has eluded efforts to understand how it promotes disease. Now, in the February 3 Proceedings of the National Academy of Sciences, researchers led by Mark Cookson at the National Institute on Aging in Bethesda, Maryland, make the case that mutated versions of the protein fuel degradation of cellular organelles through autophagy. The authors identified three proteins that form a complex with LRRK2—a large, complicated protein—and promote degradation of Golgi vesicles. Intriguingly, all pathogenic forms of LRRK2 tested in this study ramped up this degradation, no matter what effect a given variant had on LRRK2’s native kinase and GTPase activities. In addition, two of the proteins may be encoded by Parkinson’s risk genes, since they lie near genetic polymorphisms identified in genome-wide association studies of sporadic PD. The findings suggest that similar pathogenic pathways underlie familial early onset and sporadic late-onset forms of the disease. “When we talk about sporadic and familial disease, we’re probably talking about an overlapping set of mechanisms. That’s the most exciting result to us,” Cookson told Alzforum. LRRK2, aka leucine-rich repeat kinase 2 currently, occupies rank 4 on PD Gene.
Other researchers hailed the findings as important. “This paper is the first to identify proteins that directly interact with LRRK2. Another exciting thing is that all three proteins they characterized belong to the same cellular pathway, supporting the idea that the pathway is important for Parkinson’s,” said Quyen Hoang at Indiana University School of Medicine, Indianapolis, who wrote an accompanying PNAS commentary.
Previous work had suggested a role for LRRK2 in Golgi trafficking, endocytosis, and autophagy, which is the cellular process that directs unneeded structures to lysosomes for degradation (see Dec 2009 news story; Oct 2012 news story; Jun 2012 news story; and Mar 2013 news story). However, it was unclear if any of this played a part in Parkinson’s. LRRK2 contains numerous protein-protein binding domains, hence it likely interacts with many molecules, and other studies have turned up functions in diverse cell signaling pathways (see Jan 2009 news story; Oct 2012 news story). Some groups have attempted to identify direct binding partners of LRRK2 using techniques such as yeast two-hybrid screens, but without success.
Instead of using yeast screens, first author Alexandra Beilina added labeled LRRK2 to arrays containing thousands of proteins to see if LRRK2 stuck to any. The scientists discovered three binding partners: Bcl-2 associated athanogene 5 (BAG5), member Ras oncogene family-like 1 (Rab7L1), and cyclin G-associated kinase (GAK); the latter currently holds the number 6 spot on PD Gene. Beilina and colleagues verified these interactions using co-immunoprecipitation and other biochemical methods. Addition of the chaperone protein Hsc70/Hsp70 stabilized the binding of LRRK2 to BAG5 and GAK. Sequential co-immunoprecipitations then showed that all five proteins formed a single complex.
What does the complex do? To answer this, the authors first transfected mouse primary neurons with tagged versions of each of the proteins. They found that LRRK2, GAK, and Rab7L1 all localized to vesicles that bud off from the Golgi apparatus. They then performed co-transfections of these proteins and saw trans-Golgi vesicles were being depleted over 72 hours, hinting that the complex plays a role in clearing these little membrane sacs. Knockdown of any component of the complex saved the trans-Golgi vesicles. Rab7L1 appeared to play a crucial role in recruiting LRRK2 to the vesicles, as transfection with inactive Rab7L1 kept the LRRK2 complex out of these vesicles.
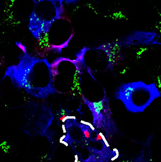
Trans-Golgi vesicles (green) disappear in cells with clustered LRRK2 (red), as seen in the outlined cell. Beilina et al., 2014. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson’s disease. PNAS.
The authors next wondered what the LRRK2 mutations might do. Testing several pathogenic variants, they found that they all increased clearance of trans-Golgi vesicles. This occurred even though the mutations affected LRRK2’s native activities in different ways: G2019S ramps up kinase activity, G2385 dampens it, while R1441C and Y1699C dial down LRRK2’s GTPase function. “We’ve been working on LRRK2 for nearly 10 years, and I have never before seen all [tested] mutations have the same effect in the same direction,” Cookson told Alzforum.
When the authors made mutants that completely lacked kinase or GTPase activity, however, they saw no change in vesicle clearance, suggesting that at least some LRRK2 activity is essential for this process. This jibes with previous studies that link LRRK2’s kinase activity to toxicity. However, LRRK2 without kinase activity seems to become less stable and its cellular levels drop (see Herzig et al., 2011). This past month, researchers led by Steven Finkbeiner at the Gladstone Institute of Neurological Disease, San Francisco, California, in collaboration with Cookson, reported that the change in LRRK2 levels alone explained the lower toxicity of kinase-dead variants (see Jan 2014 news story). While the role of LRRK2’s kinase activity in disease has been controversial, Cookson’s new study also supports the idea that loss of this function fails to directly explain toxicity.
To see if their results held up in-vivo, Cookson’s group transfected adult mouse striatum with lentivirus expressing G2019S LRRK2. As expected, they saw fewer trans-Golgi vesicles than in control mice. Blocking autophagy with pharmacological inhibitors or RNA interference rescued vesicles.
Other Parkinson’s risk factors, for example the lysosomal enzyme glucocerebrosidase (GBA, currently number 3 on PD Gene), play crucial roles in autophagy as well (see Jun 2009 news story; Jun 2012 news story). Cookson noted that parkin and Pink1 have been found to promote disposal of a different organelle, namely mitochondria, via mitophagy, a type of autophagy (see Feb 2010 news story; Aug 2013 news story). “We think LRRK2 plays a similar role in bringing its binding partners together to the right part of the cell to turn over Golgi, or at least a subset of Golgi-derived vesicles,” he said. It remains unclear, however, how enhanced clearance of trans-Golgi vesicles might lead to Parkinson’s disease. Cookson plans to investigate this by studying the long-term effects of LRRK2 mutations in the brain in-vivo.
The new data dovetail with other work implicating LRRK2 in autophagy, Cookson said. The recent Finkbeiner paper reported that high levels of wild-type or mutant LRRK2 lead to cell death through a mechanism that requires α-synuclein and seems to involve protein turnover. Cookson noted that α-synuclein has been shown to affect vesicle recycling and protein homeostasis (see Sep 2010 news story; Apr 2013 news story; Fornai et al., 2005). “There may be a similar underlying mechanism to LRRK2 and α-synuclein toxicity,” Cookson speculated.
Faulty autophagy has been linked to Alzheimer’s disease (see May 2011 news story) as well as other neurodegenerative disorders (see Aug 2011 news story), but most such studies report that the process slows, rather than accelerates. Ralph Nixon at the Nathan Kline Institute in Orangeburg, New York, pointed out that trans-Golgi vesicles deliver crucial components to lysosomes. Therefore, depletion of these vesicles might lead to a general block in autophagy, he speculated. “That would do the same sort of thing that α-synuclein and GBA do, which is to corrupt lysosomal function,” Nixon said. He suggested that future work might test this hypothesis. All neurodegenerative diseases are diseases of aging, and aging also turns down lysosome activity, he noted.
Another aspect of this new research was the identification of Rab7L1 and GAK as potential Parkinson’s genes. Both lie within regions flagged by GWAS as risk loci for sporadic PD, but the functional variants were unknown. Looking for protein interactions may represent a way to pin down other GWAS hits, Cookson suggested and commentators agreed. “I think this is very important. It tells us that cell biology can guide genetics for a change. This type of approach will now help us find other Parkinson’s genes and can be applied to other diseases as well. I am excited by the idea that we are identifying pathways to disease,” John Hardy at University College London wrote to Alzforum.—Madolyn Bowman Rogers.
References
News Citations
- α-Synuclein Conspires With LRRK2 to Corrupt Neurons
- New Substrate for Parkinson’s Protein Is Picky About Phosphate
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- LRRK2 Pathway Offers Up New Targets in Parkinson’s
- The Many Faces of LRRK2
- Rethinking LRRK2 Toxicity: It’s How Much Is Present, Not What It Does
- More Than Gaucher’s—GBA Throws Its Weight Around Lewy Body Disease
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
- α-Synuclein’s Day Job: To Chaperone SNARE Complexes?
- Synuclein Resists and Disrupts Autophagy
- Lysosomal Block Clogs Transport, Swells Neurites
- New ALS Genes Implicate Protein Degradation, Endoplasmic Reticulum
Paper Citations
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011 Nov 1;20(21):4209-23. PubMed.
- Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005 Mar 1;102(9):3413-8. PubMed.
External Citations
Further Reading
Primary Papers
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, International Parkinson’s Disease Genomics Consortium, North American Brain Expression Consortium, Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans JM, Greggio E, Cookson MR. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014 Feb 18;111(7):2626-31. Epub 2014 Feb 7 PubMed.
- Hoang QQ. Pathway for Parkinson disease. Proc Natl Acad Sci U S A. 2014 Feb 18;111(7):2402-3. Epub 2014 Feb 7 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Icahn School of Medicine at Mount Sinai
With increasing numbers of genetic risk factors for PD being identified, critical questions are whether these genes are connected, function in the same cellular pathways, or whether a common pathway exists for the majority of PD cases. Using large-scale screening, the Cookson group found three proteins that interact with LRRK2, an important PD-linked protein kinase. Two of them, Rab7L1 and GAK, were identified as risk factors for PD in previous GWAS studies. The new findings provide insight into functional connectivity of the three PD genes at the molecular level, and suggest a common pathway may be implicated in idiopathic PD. That the same “hits” arise from two completely independent approaches is particularly interesting and extremely valuable in the validation of pathogenic pathways in PD. In fact, the results from Cookson’s group, together with the previous evidence provided by Abeliovich and colleagues for the genetic interaction between Rab7L1 and LRRK2 (MacLeod 2013), open new avenues for the research of pathogenic pathways in more common forms of PD.
View all comments by Zhenyu YueMake a Comment
To make a comment you must login or register.