Immune Receptor Binds Aβ Oligomers, Spurs Synaptic Loss
Quick Links
The list of claimed Aβ receptors is growing, and the latest addition hails from the immune system. In the September 20 Science, Carla Shatz of Stanford University, Palo Alto, California, and collaborators elsewhere report Aβ oligomer binding by PirB, a mouse immunoglobulin-like receptor expressed on neurons, as well as by its human ortholog, the leukocyte immunoglobulin-like receptor LilrB2. Deletion of the mouse receptor—paired immunoglobulin-like receptor B—in transgenic mice modeling Alzheimer’s disease enabled the animals to resist synaptic plasticity loss and behavioral deficits typically triggered by amyloid-beta. Furthermore, Aβ binding to the immune receptors aberrantly boosted signaling through the actin cytoskeleton.
“If Aβ neurotoxicity is indeed caused by specific receptor-ligand interactions, then the neuroimmune receptor PirB is clearly a candidate for mediating at least some of the disease symptoms,” Iryna Benilova and Bart De Strooper, University of Leuven, Belgium, wrote in a commentary on the new study in the same issue of Science.
PirB (see image below) came to Shatz’ attention through her longstanding interest in neural development. More than a decade ago, she and colleagues discovered that mice lacking class I major histocompatibility (MHC) proteins—the ligands for T-cell receptors—develop abnormally due to exaggerated plasticity and faulty synapse removal (Huh et al., 2000). The scientists identified PirB as an MHC class I receptor, and showed that PirB-deficient mice had a similar phenotype (Syken et al., 2006). The Shatz lab studies synaptic plasticity in the visual system by measuring ocular dominance plasticity (ODP), the process by which brain circuits reallocate connections to an open eye if the other is closed or damaged during development. Loss of PirB leads to exaggerated ODP in wild-type mice.
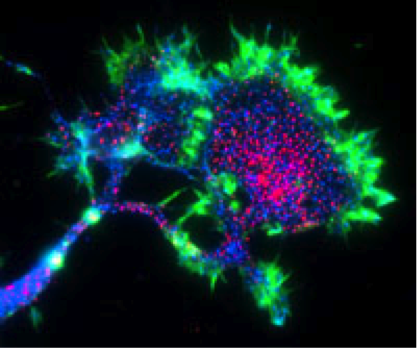
Another Aβ receptor? The neuroimmune receptor PirB (red) is heavily concentrated on the growth cone of mouse cortical neurons. Image credit: Carla Shatz and Josh Syken, Stanford University.
How might this relate to neurodegeneration? “If AD starts way before plaques and tangles appear, and is a disease of synapses,” Shatz told Alzforum, “then maybe we would see ODP defects in AD mice early on.” Lo and behold, two strains—the APP/PS1 and Tg2576 transgenics—showed none of the signs of ODP that wild-type mice do when tested at 4-5 weeks of age (ARF news story). That work—done by co-author Chris William in Brad Hyman’s lab at Massachusetts General Hospital in Boston—suggested to Shatz that synaptic plasticity falters well in advance of Aβ pathology and symptoms. Given that PirB knockout mice had too much plasticity while AD mice had too little, could removing PirB restore ODP to the visual cortex of AD mice? “One way this could work is if PirB actually binds beta-amyloid,” said Shatz, presuming that Aβ might hijack the PirB receptor away from its normal partners.
Both hunches proved true, first author Taeho Kim and colleagues report in this week’s paper. APP/PS1 mice bred onto a PirB-deficient background had normal ODP. Immunostaining and immunoprecipitation experiments with PirB-overexpressing HEK293 cells and mouse cortical neurons showed synthetic Aβ42 oligomers binding PirB with nanomolar affinity. Larger species appeared to bind PirB more so than monomers, dimers, or trimers, though the paper did not provide a detailed characterization of the oligomer species at play (see Alzforum webinar.)
Neurons from PirB knockout mice bound Aβ42 oligomers about half as well as wild-type neurons, indicating that Aβ binding is partially PirB-dependent. However, the incomplete loss of binding also suggests PirB is not the lone receptor for Aβ oligomers. Consistent with that, others have reported that Aβ binds NMDA receptors (ARF news story on Snyder et al., 2005), cellular prion protein (ARF conference story), the receptor tyrosine kinase EphB2 (ARF news story), the receptor for advanced glycation end products, aka RAGE (ARF news story), metabotropic glutamate receptor 5 (ARF news story), and the immune cell receptor FcγRIIb (ARF news story).
Importantly, the present study also showed that Aβ-PirB association disrupts neuronal function. When the researchers dumped Aβ onto hippocampal slices from mice with and without PirB, they found that PirB-deficient slices were immune to the loss of synaptic strengthening (long-term potentiation) Aβ normally inflicts on wild-type brain tissue. Moreover, nine-month-old mice growing up without PirB performed as well as wild-type mice on recognition memory tasks, Shatz said.
As a step toward testing the relevance of this finding in humans, Shatz’ team found that Aβ42 oligomers also bind the human homolog of PirB—leukocyte immunoglobulin-like receptor B (LilrB2). Western blotting detected LilrB2 protein on postmortem brain specimens from both healthy elderly and people with AD. However, downstream signaling looked different in brain extracts from APP/PS1 mice and AD patients, relative to their respective controls. When Aβ is around, PirB interacts more with protein phosphatases PP2A, PP2B (calcineurin), PP2C, and cofilin, an actin-depolymerizing factor involved in memory consolidation. AD brains have an abundance of cofilin (ARF news story), a protein thought to regulate dendritic spine morphology and cellular memory correlates (Rust et al., 2010). Previous work in the Hyman lab suggests that Aβ peptides cause neurons to shrivel and lose spines in part by activating calcineurin (ARF news story).
All told, the findings expand the list of putative Aβ receptors but also raise questions about the critical features of these associations. “Do the numerous ‘Aβ receptors’ simply reflect the sticky nature of Aβ oligomeric species, or is there a particular ‘pathological conformation’ in some of the putative oligomeric intermediates that promotes interaction with specific receptors?” Benilova and De Strooper ask in their editorial.
Moustapha Cissé of INSERM in Valbonne, France, notes that the use of synthetic (i.e. non-physiological) Aβ oligomers has been known to skew interactions with endogenous proteins and suggested confirming the results with Aβ oligomers derived from AD brain or CHO cells (see full comment below). In the end, “the key question is not so much whether sticky Aβ oligomers bind a particular receptor, but whether such Aβ receptor-mediated pathways are functionally meaningful and can ultimately be used to create novel therapies,” Benilova and De Strooper wrote. Toward that end, Shatz and colleagues have designed a PirB inhibitor and plan to test if it can boost synaptic plasticity in the brains of adult wild-type mice and AD models. To Shatz’ knowledge, LilrB2 gene variants have not cropped up in AD genomewide association studies. LilrB2 sits near apolipoprotein E (the top AD risk gene) on chromosome 19.—Esther Landhuis
References
News Citations
- Synaptic Plasticity Falters Early in AD Mice
- Amyloid-β Zaps Synapses by Downregulating Glutamate Receptors
- Keystone: Partners in Crime—Do Aβ and Prion Protein Pummel Plasticity?
- Aβ Downs EphB2 Kinase, Disrupts Glutamate Receptors
- Aβ Oligomers and Synaptic Dysfunction—Blame It on RAGE?
- Glutamate Receptor Links Aβ-Prion Complex with Fyn, Synaptic Damage
- Immune Cell Receptor Gives Aβ a Toxic Edge
- AD Pathology—Loss of Kinase Sends Synapses PAKing
- Calcium Hypothesis—Studies Beef Up NFAT, CaN, Astrocyte Connections
Webinar Citations
Paper Citations
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000 Dec 15;290(5499):2155-9. PubMed.
- Syken J, GrandPre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006 Sep 22;313(5794):1795-800. PubMed.
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005 Aug;8(8):1051-8. PubMed.
- Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Banchaabouchi MA, Giustetto M, Triller A, Choquet D, Witke W. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010 Jun 2;29(11):1889-902. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005 Aug;8(8):1051-8. PubMed.
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000 Dec 15;290(5499):2155-9. PubMed.
Primary Papers
- Benilova I, De Strooper B. Neuroscience. Promiscuous Alzheimer's amyloid: yet another partner. Science. 2013 Sep 20;341(6152):1354-5. PubMed.
- Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science. 2013 Sep 20;341(6152):1399-404. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Gladstone Institutes/UCSF
Aβ oligomers bind to multiple proteins and have diverse effects in cell culture and animal models of the disease, depending on a plethora of variables such as oligomer species, length of exposure of neurons to oligomers, mutations harbored by transgenic mice, type of transgenes, etc... Mechanisms by which Aβ oligomers cause neuronal network dysfunction have been sparse in the literature, and many groups in the field of AD research focus on trying to understand these mechanisms. The study by Kim & al. identifies PirB and its human ortholog LilrB2 as receptors for Aβ oligomers. Thus, this study expands the list of proteins that have been identified as key mediators of Aβ oligomer-dependent toxicity. The authors used a wide range of approaches, including biochemical assays, electrophysiology and behavior that were well designed to provide convincing clues and new leads on how these toxic species impair cognitive functions and elicit memory impairments in an AD model.
The authors identify the two most N-terminal Ig domains of PirB and LilrB2 as critical for interaction with high-n Aβ oligomers as deletion of these domains significantly abrogates this interaction. It is interesting to note that low-n oligomers (dimers and trimers) were not pulled down with PirB-Fc or LilrB2-Fc. This raises the possibility of a receptor selectivity based on Aβ oligomers species. However, the authors did not clearly identify the specific oligomeric Aβ involved in this interaction. In this study for instance, could Aβ*56 be the main oligomer species causing the neuronal network dysfunction in the APP/PS1 line? Another important point is that the authors did not test other sources of Aβ oligomers (i.e. CHO-derived or AD brain-extracted Aβ oligomers) that have been shown to be more potent than synthetic and might be less prone to artificial interaction.
The authors also show that Aβ oligomer binding to neuronal cells was not fully abolished in the absence of PirB, meaning that additional receptors exist. This is not surprising in light of the literature and the growing number of proteins that have been shown to bind Aβ oligomers. Aβ oligomers affect many signaling pathways to cause spine loss and cognitive deficits, it is therefore tempting to speculate that normalizing/fixing only one of these different pathways might be sufficient to alleviate or prevent these deficits.
The authors also established that the PirB/oligomer interaction exacerbates cofilin signaling leading to dendritic spine loss and cognitive impairment. It would be very informative to examine A oligomer-induced dendritic spine loss by cell imaging and determine whether this is prevented by PirB removal in PirB-/tg mice or in cell culture.
It would also be very interesting in future studies to assess whether the PirB/oligomer interaction is present in other AD models and leads to impairment of cofilin signaling and subsequent spine loss. Although the authors assessed the rescue of cognitive deficits by two different tests (NOR and NPR), testing PirB-/tg mice in the Morris Water Maze in a follow-up study would also provide additional information.
Overall, this is a great study that contributes to a better understanding of the mechanisms by which Aβ oligomers harm the brain and opens up new leads in AD research.
Cognition Therapeutics Inc.
This is a very important manuscript demonstrating that Aβ oligomers bind to and adversely affect a molecule critical for normal learning and memory, adding to the evidence that Alzheimer’s begins and persists as a synaptic plasticity disease. It was discovered through an unbiased screen, and is another example of how this approach is proving fruitful for identifying molecular mechanisms involved in the disease process. The identification of a role for PirB as an oligomer receptor is a breakthrough for the field, and provides further support for efforts to discover and develop therapeutics that antagonize these early binding and signaling events. It holds the promise of being able to intervene in the Alzheimer's disease process, even after diagnosis, by directly blocking the effects of soluble Aβ oligomers on synapse function.
The accompanying preface to the article points out that the exact structure of Aβ oligomers that cause toxicity is still unknown, and many labs work with unstandardized heterogenous mixtures of different oligomer structures. It suggests that these unstandardized mixtures may be one reason behind the identification of several putative oligomer receptors. While it is certainly urgent that action be taken towards structural understanding of disease-relevant oligomers, an alternative interpretation is possible. Rather than exist as separate entities as is depicted in the accompanying diagram, several of these candidate receptor molecules may be closely linked in a multi-protein receptor complex, whose specific members may change as a result of electrophysiological activity or damage. Additionally, such multi-protein receptor complexes need not be exclusively postsynaptic. Indeed, molecules on both sides of the synapse are in direct contact, and this is critically required for normal function. This paper has identified a key presynaptic player in this emerging story.
The interactions that cause Alzheimer’s disease may prove to be more tractable than complex oligomer ligands and multiple receptors would currently lead us to believe. Pathological processes that behave according to pharmacological principles contain signaling nodes that are possible to target with therapeutics. It is now quite urgent to confirm that this is the case in Alzheimer’s disease. For the sake of patients and their families, let us hope so.
Make a Comment
To make a comment you must login or register.