Imaging Reveals Amyloid Up To a Year After Traumatic Brain Injury
Quick Links
For more than 20 years, scientists have known that a blow to the head triggers rapid accumulation of Aβ in the brain, the same peptide that builds up slowly in people developing Alzheimer’s disease (see Gentleman et al., 1997). Researchers now report that a tracer used to detect amyloid in AD patients picks up Aβ plaques in victims of traumatic brain injury (TBI) as well. In a cross-sectional study, scientists led by David Menon, University of Cambridge, England, found that the imaging agent Pittsburgh compound B (PiB) bound the cortex and striatum of TBI patients. In the future, such tracers will allow researchers to longitudinally observe the time course of this buildup, probe whether and how fast it clears, and discover any links to cognitive impairment, Menon told Alzforum. The report appeared in the November 11 JAMA Neurology.
Until now, scientists have relied on biopsy and postmortem data to study Aβ in TBI. Plaques appeared to arise within hours of injury and crop up in about a third of patients (see Roberts et al., 1994). Some studies hinted that the plaques cleared after a time (see Chen at al., 2009), while others found that it stuck around long-term (see Johnson et al., 2012). Other studies suggested that TBI heightened the risk for dementia later in life (see Aug 2012 news story). However, the nature of brain injuries varies widely, and scientists caution against making inferences about the extent or long-term implications of amyloid in TBI without longitudinal data. Recently developed amyloid imaging agents for positron emission tomography have allowed scientists to visualize Alzheimer’s-related plaques in people. Menon’s group wanted to explore whether that same technology could be applied to TBI patients.
First author Young Hong and colleagues used PiB to image amyloid in the brains of 15 people, aged 21 to 50 years, who in the past year had survived a catastrophic traumatic brain injury, such as that resulting from a car crash. Eleven healthy people, aged 24 to 60, served as controls. Analysis of a separate cohort of 16 people who died following a TBI confirmed that PiB bound to Aβ plaques detected in postmortem immunohistochemistry of brain tissue. Numerous studies have reported that PiB binding specifically correlates with amyloid plaques in AD patients postmortem (see Mar 2008 news story on Ikonomovic et al., 2008; Driscoll et al., 2012).
Compared with the healthy group, more PiB bound in the cortical gray matter and striata of the 15 TBI survivors (see image below). Levels of PiB binding occasionally equaled those of people with mild cognitive impairment, who often go on to develop Alzheimer’s. In one case, binding exceeded that typical for AD. Interestingly, in many of the TBI patients, PiB strongly bound in the striatum, resembling a pattern seen in early onset AD patients with mutations in the presenilin-1 (PS1) gene (see Jun 2007 news story). This contrasts with late-onset AD, where the plaques appear primarily in the cortices and later in the hippocampus (see Braak and Braak, 1991). Because PS1 mutations cause overproduction of Aβ, the same mechanism could underlie amyloid buildup in TBI, Menon told Alzforum. How? When damaged axons no longer traffic the amyloid precursor protein properly, more gets cleaved by β- and γ-secretase, leading to elevated Aβ, he proposed. “That’s the essential new clue to how and when we might intervene in TBI patients,” he added.
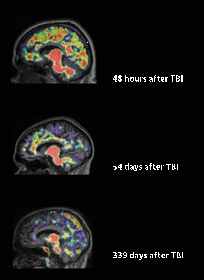
Amyloid plaques in a cross-section of TBI patients. American Medical Association. Copyright © (2013). All rights reserved.
The patients tested had suffered a TBI between two and 349 days prior to the PiB scan. While the data hint that post-TBI amyloid in the brain clears with time, Menon cautions that no conclusions can be drawn by comparing patients who had head injuries of differing severity. Only serial images will shed light on that hypothesis, he said. Amyloid imaging will allow us to do just that, he added. Longitudinal imaging will let scientists estimate what proportion of patients accumulate amyloid and whether they have a higher risk for AD later on, determine how cognitive function tracks with amyloid levels, and find out if some factors—such as ApoE status—affect build up. Postmortem studies suggest that people who carry the ApoE4 allele fare worse than noncarriers after a brain injury (see Zhou et al., 2008). Amyloid imaging also may help researchers determine whether better amyloid clearance leads to more complete recovery, Menon said. Steven DeKosky, University of Virginia, Charlottesville, agreed. “Eventually, if upregulation of amyloid predicts a worse outcome, then researchers might conduct a trial to determine if removing Aβ with an antibody or an enzyme inhibitor would lead to a better outcome,” he said. Such trials could provide insights about the effectiveness of amyloid removal in AD as well, said Menon.
Menon plans to follow these 15 volunteers to see if their cognitive performance improves or declines. He also wants to test if amyloid deposition correlates with structural brain changes.
Though amyloid imaging represents an improvement over postmortem tissue in the study of TBI, it still may not capture the whole picture, said Samuel Gandy, Mount Sinai School of Medicine, New York. He pointed out that many of the plaques seen in TBI are diffuse, and invisible to current ligands. For that reason, this method may underestimate the true extent of proteinopathy.—Gwyneth Dickey Zakaib
References
News Citations
- New AlzRisk Analysis: Brain Injury Promotes Dementia, But Is It AD?
- Amyloid Imaging: Laying PIB Concerns to Rest
- eFAD Research Surprise: In Mutation Carriers, Amyloid Starts in Striatum
Paper Citations
- Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin WS, Graham DI. A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport. 1997 Apr 14;8(6):1519-22. PubMed.
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994 Apr;57(4):419-25. PubMed.
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009 Apr;19(2):214-23. PubMed.
- Johnson VE, Stewart W, Smith DH. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol. 2011 Jun 29; PubMed.
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, Dekosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008 Jun;131(Pt 6):1630-45. PubMed.
- Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, Kraut MA, Ferrucci L, Mathis CA, Klunk WE, O'Brien RJ, Davatzikos C, Wong DF, Resnick SM. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol. 2012 Aug 5; PubMed.
- Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008 Apr;25(4):279-90. PubMed.
Other Citations
Further Reading
Papers
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013 Jan;136(Pt 1):28-42. PubMed.
- Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008 Apr;25(4):279-90. PubMed.
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994 Apr;57(4):419-25. PubMed.
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009 Apr;19(2):214-23. PubMed.
- Johnson VE, Stewart W, Smith DH. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol. 2011 Jun 29; PubMed.
News
- Stress and Trauma: New Frontier Lures Alzheimer’s Researchers
- Dementia Four Times More Likely in Pro Football Players
- Blast Anatomy—Chronic Traumatic Encephalopathy in Military Vets
- Keystone: Metabolic and Axonal Dysfunction in Traumatic Brain Injury
- Keystone: Traumatic Brain Injury—Epidemiology and Characteristics
Primary Papers
- Hong YT, Veenith T, Dewar D, Outtrim JG, Mani V, Williams C, Pimlott S, Hutchinson PJ, Tavares A, Canales R, Mathis CA, Klunk WE, Aigbirhio FI, Coles JP, Baron JC, Pickard JD, Fryer TD, Stewart W, Menon DK. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014 Jan;71(1):23-31. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.