Genetic Risk Score Combines AD GWAS Hits, Predicts Onset
Quick Links
In lieu of a crystal ball that can foresee Alzheimer’s, scientists are trying to divine impending disease by looking deep into a person’s genome. As reported in the March 21 PLoS Medicine, scientists led by Rahul Desikan of the University of California, San Francisco, Ole Andreassen of the University of Oslo, Norway, and Anders Dale of UC San Diego have formulated a genetic score that predicts how old people will be when they develop Alzheimer’s disease. The model considers everyone at risk. This “polygenic hazard score” (PHS) combines the risk from common AD-associated genetic variants into a single value. In various independent populations, the PHS predicted age at onset and rate of progression to AD, and it correlated with both in vivo biomarkers and neuropathology. The PHS could help enrich clinical trial samples and improve differential diagnosis, said the authors.
“The analysis is incredibly thorough,” said Elizabeth Mormino, Stanford University School of Medicine, California, who was not involved in the research. “This is the first paper to take such a wide-scale approach to validating a polygenic risk score in the context of Alzheimer’s disease.” Mormino has calculated a polygenic score that correlated with cognitive and structural brain changes that precede AD (see Jul 2016 news).
On their own, single nucleotide polymorphisms (SNPs) identified in genome-wide association studies (GWAS) usually raise the risk of disease only very slightly. Together, however, risk from other SNPs may add up. To calculate a person’s total genetic burden, scientists have started pooling SNP data into polygenic risk scores (Chouraki et al., 2016). They devise these scores by comparing genotypes of cases to genotypes of controls who have not developed AD. The problem is, some of those controls may develop disease later, said co-author James Brewer, UC San Diego.
Dale, Desikan, and co-first author Chun Chien Fan, UC San Diego, decided to model AD a different way. They developed a polygenic “hazard” score that assumed everyone would eventually develop the disease, with the only question being when. To start off, they first compared 17,000 AD cases to more than 37,000 controls from the International Genomics of Alzheimer’s Project and chose 1,854 SNPs that appeared more often among AD patients, even though most of them fell below the threshold for genome-wide significance. They then examined these SNPs in 6,400 AD cases and 9,386 controls from the first phase of the Alzheimer’s Disease Genetics Consortium (ADGC) and determined the average age at onset associated with each SNP. They incorporated the riskiest SNPs one by one into their model, ultimately picking 31—including many of the known AD risk alleles from past GWAS—that together best predicted age at onset. By comparing the hazard associated with each of these 31 SNPs with age-related incidence rates from the general population, the authors derived a PHS for each person.
Before taking the PHS for a real-world whirl, the authors checked to make sure it added predictive value above that of ApoE4, which alone increases the chance of getting AD by 15-fold in E4 homozygotes. They analyzed 4,969 ApoE3/3 homozygotes within the ADGC. The PHS score successfully stratified volunteers by age of onset over the 11-year range in this cohort. Those with the highest PHS scores developed the disease at around age 84, while those with the lowest scores were diagnosed around age 95.
To test the score further, the authors then looked at 6,900 AD cases from Phase 2 of the ADGC. Once again, people with a higher PHS got the disease earlier than those with a lower score, and the predicted age at onset closely matched the actual age (see images below). 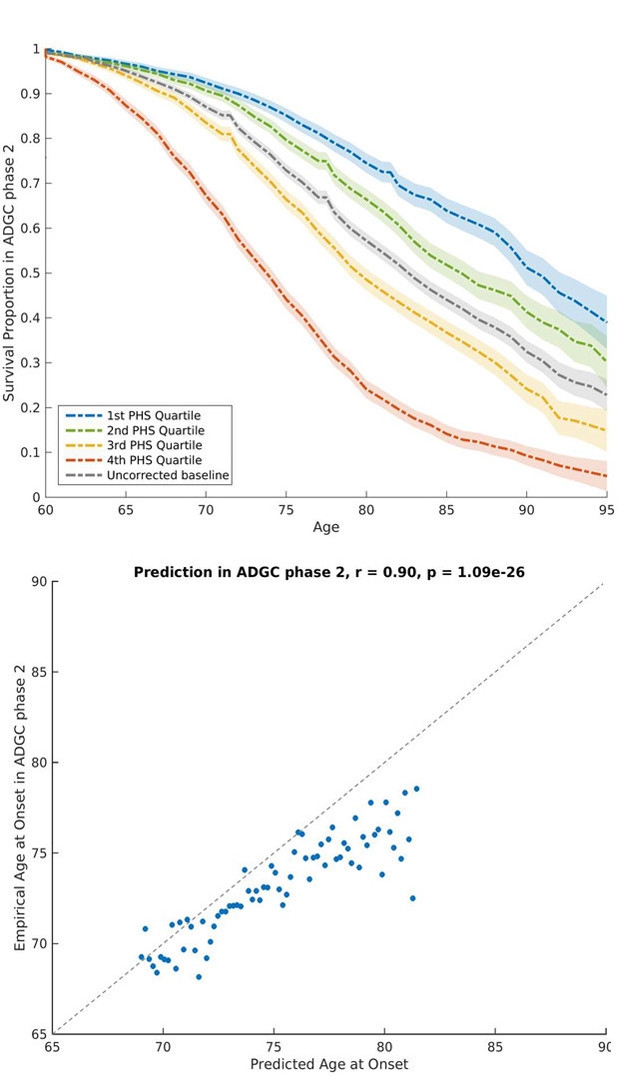
Hazard Score. At top, people in the quartile with the lowest PHS (blue) live AD-free longer than those with the highest score (red). At bottom, the PHS-predicted and actual age at onset closely agree. [Courtesy of Desikan et al. 2017.]
The PHS score also associated with other AD-related phenotypes. In a sample population from the National Institute on Aging Alzheimer’s Disease Center (NIA ADC) of 2,724 cognitively normal people who were followed for at least two years, high scorers progressed to AD faster than people with low scores. Those with high scores also had more plaques and tangles at autopsy. PHS correlated with in vivo biomarkers and cognition, too. Of 692 people in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a high PHS score correlated with lower Aβ42 and higher total tau in the cerebrospinal fluid, as well as a lower volume of medial temporal lobe structures and a weaker score on the CDR Sum of Boxes.
Together, the data lend credence to the idea that these AD-related phenotypes have a genetic component, said Carlos Cruchaga, Washington University in St. Louis, who was not involved in the study. “Our genetic knowledge has increased a lot in the past five to 10 years,” he said. “Now we are at a point where we can combine all these genetic analyses to get better at predicting individual risk of onset and progression.” Models will become even more reliable when they incorporate more than just common variants, he added. “Once we integrate rare variants, as well as biochemical and imaging biomarkers, that’s when we will have a pretty good prediction model.”
Used in a research setting, the PHS could help enrich clinical trial samples by allowing researchers to select participants expected to decline fastest, said Brewer. At less than $100, it could also be a cheap way to screen for people most likely to test positive for amyloid or tau in a PET scan, he said.
The authors are still assessing how the PHS could be used clinically. Brewer speculates that it could aid in differential diagnosis, helping distinguish AD from other dementias. It could also improve the family history assessment, which clinicians often consider when making a diagnosis; rather than just asking whether a relative developed the disease, a clinician can objectively assess a person’s genetic background. Polygenic risk scores are already proving useful in breast cancer prevention by helping physicians determine when women should start mammogram screening, and in cardiovascular disease, determine who will benefit from statins. It’s a different story in those fields because there are treatments for those diseases, said Brewer. However, if and when one becomes available for Alzheimer’s, this type of score will be valuable for deciding who will benefit, he told Alzforum.
These types of studies demonstrate that AD has a strong genetic component beyond ApoE, and this is extremely useful in predicting and modifying AD risk, said Valentina Escott-Price, Cardiff University, Wales. She added that these findings still need to be replicated in larger independent samples. Fan noted that the need for large sample sizes meant they had to restrict their analysis to non-Hispanic whites of European descent. Future studies should sample from multiple ethnic groups, he said.
A related paper in the February 17 Neurology suggested that a polygenic risk score has predictive value regardless of ethnic group. Richard Mayeux and Giuseppe Tosto, Columbia University, New York, reported that in non-Hispanic whites of European and North American descent in the NIA Late-Onset Alzheimer’s Disease Family Study cohort, an uptick in genetic risk score equivalent to one standard deviation increased the chances of developing AD by 29 percent, and lowered age at onset by eight months. They calculated a similar polygenic risk score for Caribbean Hispanic families from the Dominican Republic and New York who were affected by LOAD. Each standard deviation increase raised the risk of LOAD by 1.73-fold.—Gwyneth Dickey Zakaib
References
News Citations
Paper Citations
- Chouraki V, Reitz C, Maury F, Bis JC, Bellenguez C, Yu L, Jakobsdottir J, Mukherjee S, Adams HH, Choi SH, Larson EB, Fitzpatrick A, Uitterlinden AG, de Jager PL, Hofman A, Gudnason V, Vardarajan B, Ibrahim-Verbaas C, van der Lee SJ, Lopez O, Dartigues JF, Berr C, Amouyel P, Bennett DA, van Duijn C, DeStefano AL, Launer LJ, Ikram MA, Crane PK, Lambert JC, Mayeux R, Seshadri S, International Genomics of Alzheimer’s Project. Evaluation of a Genetic Risk Score to Improve Risk Prediction for Alzheimer's Disease. J Alzheimers Dis. 2016 Jun 18;53(3):921-32. PubMed.
Further Reading
Papers
- Sims R, Williams J. Defining the Genetic Architecture of Alzheimer's Disease: Where Next. Neurodegener Dis. 2016;16(1-2):6-11. Epub 2015 Nov 10 PubMed.
Primary Papers
- Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, Thompson WK, Besser L, Kukull WA, Holland D, Chen CH, Brewer JB, Karow DS, Kauppi K, Witoelar A, Karch CM, Bonham LW, Yokoyama JS, Rosen HJ, Miller BL, Dillon WP, Wilson DM, Hess CP, Pericak-Vance M, Haines JL, Farrer LA, Mayeux R, Hardy J, Goate AM, Hyman BT, Schellenberg GD, McEvoy LK, Andreassen OA, Dale AM. Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Med. 2017 Mar;14(3):e1002258. Epub 2017 Mar 21 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Over the past few days, I have received personal emails from individuals asking about getting our polygenic hazard score (PHS) for themselves and physicians asking about clinical availability of this test for their patients. On the other hand, in several news stories, I have seen comments from researchers and clinicians stating that our PHS "is nowhere near ready for clinical use" and needs "additional validation in large independent cohorts."
As a practicing clinician, my main concern would be improper use and interpretation of our new PHS. However, I think it is important to better understand whether, and in what context, our PHS can be currently used, what next steps are needed for broad clinical use, and to dispel any false claims about usability/non-usability of our test.
First, our research study likely represents one of the largest genetic studies ever conducted in the United States, and we used all available genetic data from U.S.-based individuals of European descent. Importantly, we developed our PHS in one sample and independently replicated our score in three independent samples (replication n > 20,000 individuals). Although our findings need replication in non-U.S., non-Caucasian populations (see below), a blanket statement that "these findings still need to be replicated in larger independent samples" is incorrect and misleading.
Second, perhaps the most important issue for clinical use is prospective validation. That is, can you use our genetic score prospectively in non-demented older individuals to predict cognitive and clinical decline? We have just completed our follow-up study suggesting that beyond APOE, we can indeed use our PHS to prospectively identify non-demented older folks (from U.S.-based memory clinics) who decline cognitively and clinically over time.
Third, another important point is the need for evaluating our score in community-based samples. As we mention in our paper, our PHS was developed and validated on individuals from specialized memory clinics from the U.S. Given that folks from specialized clinics have faster rates of progression to AD than older folks from the general community, our score needs to be validated on a prospective sample from the general community. We are in the process of working on this.
Finally, we need to replicate our findings on non-U.S., non-Caucasian cohorts. For non-U.S. cohorts, we actually have preliminary evidence suggesting that our PHS works well at predicting Alzheimer's age of onset in older individuals from the general population from Iceland and Norway. For non-Caucasian populations, this is much harder to do as we currently do not have access to large U.S. or non-U.S. studies that have genotype, Alzheimer's age of onset, and clinical decline data.
In conclusion, our PHS right now is primarily intended for research and clinical trial use. However, given the results within our PLOS Medicine paper and our recent findings in using PHS to identify non-demented older individuals at greatest risk for clinical decline, subtitles and statements such as "Warning: False health news?'" and "we're so far away from a test that can be used clinically" (Mar 26 CBC news) are misleading and incorrect.
Bottom line, if you are an individual who is similar in make-up to the older, U.S.-based, Caucasian participants of European descent we evaluated in our study, we are working on options for offering PHS scores for Alzheimer's disease in clinical routine, and through executive health clinics.
Make a Comment
To make a comment you must login or register.