Does Presenilin 1 Moonlight as Autophagy Driver, Paradoxically Reducing Aβ?
Quick Links
Presenilin 1 is established as the catalytic subunit within γ-secretase—the enzyme conglomerate that unleashes the Aβ peptide from its precursor, the amyloid precursor protein. However, according to a pair of papers in the May 22 Proceedings of the National Academy of Sciences online, presenilin may at the same time undercut this well-proven effect. Researchers led by Paul Greengard at Rockefeller University in New York report that when phosphorylated at serine-367, PS1 teams up with a gang of vesicular proteins to kick autophagosome-lysosome fusion into high gear. This promotes the destruction of βCTF, the fragment left after β-secretase cleaves APP, thus thwarting the production of Aβ. The researchers believe that ramping up PS1 phosphorylation at serine-367 might become a therapeutic strategy to reduce Aβ overproduction.
“These are very interesting findings that show a new way that PS1 can affect Aβ levels, a pathway that may provide new potential targets for the discovery of AD therapeutics,” commented Michael Wolfe of Kansas University in Lawrence. Other researchers remained cautious in their interpretation until the subcellular pools of APP that might be subject to this bidirectional, PS1-mediated processing are further defined (see, e.g., Wim Annaert comment below).

Leading a Double Life? A study claims that besides generating Aβ, PS1 also reduces it by promoting autophagy. [Courtesy of Bustos et al., PNAS, 2017.]
This is not the first time researchers have proposed a non-catalytic role for PS1; for example, it reportedly acts as a release valve for intracellular calcium stores (Jun 2010 news). Whether or how non-catalytic functions are regulated remains a mystery, but it is possible that phosphorylation of the protein could play a role. In support of this idea, a previous study reported that phosphorylation on any of 15 sites, including serine-367, had no effect on PS1’s catalytic function (see Matz et al., 2015). However, a more recent report claimed that phosphorylation at serine-367 did affect PS1’s catalytic function, stating that phosphomimetic mutations at residues including at serine-367 forced PS1 into a “closed conformation” (Maesako et al., 2017). This conformation previously had been reported to boost the Aβ42/Aβ40 ratio, and is the shape taken by PS1 harboring familial AD mutations (Uemura et al., 2009; Berezovska et al., 2005).
Against this complex backdrop, first author Victor Bustos and colleagues set out to understand how serine-367 phosphorylation affected PS1 function. Computational analysis had predicted that serine-367 would fit the bill as a casein kinase 1 (CK1) substrate (Xue et al., 2008). Bustos knocked down each of the six CK1 isoforms in neuroblastoma cells and zeroed in on CK1γ2 as the likely kinase. Inhibiting this kinase reduced PS1 serine-367 phosphorylation by nearly 60 percent, while overexpressing CK1γ2 in mouse fibroblasts ramped it up. More intriguingly, Bustos noticed that blocking the kinase in neuroblastoma cells overexpressing APP led to a glut of Aβ peptide. That suggested CK1γ2 phosphorylation of serine-367 limited Aγ production.
To test if that would happen in vivo, the researchers generated knock-in mice, swapping endogenous PS1 with one that had alanine at residue 367 (S367A), i.e., cannot be phosphorylated there by CK1γ2. They crossed these animals to J20 mice, which overexpress APP with the Swedish familial AD mutation. Nine months later, the researchers found that levels of soluble and insoluble Aβ had skyrocketed in the offspring expressing the S367A PS1. Compared to J20 mice, the S367A-PS1/J20 mice had 40-fold more insoluble Aβ40, and a fivefold heavier plaque burden. Surprisingly, J20 animals expressing an expected phosphomimetic version of PS1, S367D, accumulated similar amounts of Aβ as those expressing the S367A version, indicating that the mimetic behaved similarly to the non-phosphorylated version.

Role Reversal. Mice expressing one (center) or two (right) copies of the non-phosphorylatable S367A-PS1 mutant have more plaques than J20 controls (left). [Courtesy of Bustos et al., PNAS 2017.]
Does serine-367 phosphorylation suppress γ-secretase activity? To answer this, the authors mixed recombinant βCTF, i.e., the substrate, with cell membrane fractions isolated from brains of wild-type or S367A mice as sources of γ-secretase. Fractions from either mouse—which the researchers confirmed contained the same amount of enzyme—produced the same amount of Aβ40 for each unit of substrate, suggesting they were equally active. Furthermore, a photoactivatable probe that only binds to catalytically active PS1 latched onto extracts from wild-type or S376A mouse brains in equal measure.
J20 mice expressing the S367A mutant harbored much more βCTF in the brain than did J20 controls, but levels of soluble APPβ, αCTF, and full-length APP were similar between the two strains. Using an antibody the authors reported to be specific for PS1 phosphorylated at serine-367, they determined that 65 percent of PS1 was phosphorylated at this residue in wild-type brain.
These findings led the researchers to hypothesize that phosphorylation of serine-367 in PS1 somehow hastened degradation of βCTF, independently of γ-secretase function. Using pulse-chase experiments to track βCTF in mouse embryonic fibroblasts, they estimated that the half-life of βCTF increased from 50 minutes in wild-type to 150 minutes in cells derived from S367A-PS1 knock-in mice.
How might serine-367 phosphorylation of PS1 fuel the destruction of βCTF? The researchers checked proteasomal processing and autophagy, finding that blocking autophagy, but not the proteasome, boosted βCTF levels in fibroblasts from wild-type mice. In cells from S367A-PS1 mice, the autophagy inhibitors did not alter βCTF levels. In both fibroblasts and cortical neurons from S367A-PS1 mice, the researchers observed increases in the autophagy markers LC3-II and SQSTM/p62, suggesting that autophagic flux had slowed down.
In their second paper, the researchers investigated how phosphorylation of PS1 might promote autophagy. Reasoning that the modification might trigger an interaction with other proteins, Bustos and colleagues started by fishing for associates. Adding phosphorylated or non-phosphorylated peptides of PS1 as bait, the researchers immunoprecipitated protein from whole mouse brain extracts. Annexin A2 specifically associated with the PS1 peptide only when it was phosphorylated at serine-367. The researchers also found that endogenous phosphorylated PS1 and Annexin A2 co-immunoprecipitated from whole brain extracts. Annexin A2 did not associate with the phosphomimetic mutant of PS1, S367D.
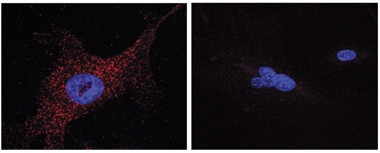
Autophagic Association.
Annexin A2 (red) associates with PS1, but not the S367A phospho-null mutant. [Courtesy of Bustos et al., PNAS 2017.]
Annexin A2 reportedly plays a role in the early stages of autophagy. The researchers used a range of biochemical techniques to piece together the relationship between PS1, Annexin A2, and autophagy. In a nutshell, they report that when phosphorylated at serine-367, PS1 associates with the N-terminus of Annexin A2 along with p11, another protein involved in endosomal trafficking. Knocking down Annexin A2 and p11, or Annexin A2 alone, slowed the flux of autophagy and dramatically boosted Aβ in neuroblastoma cells expressing APP. Using knockout mice and cell lines, the authors went on to find that the p-PS1/Annexin A2/p11 complex facilitated autophagosome-lysosome fusion by interacting with VAMP8, a lysosomal SNARE protein. Somehow this interaction pushed VAMP8 into linking up with Stx7, a corresponding SNARE on autophagosomes.
Overall, the findings point to a new function for PS1: that of autophagy driver. That said, Bustos told Alzforum that PS1 does not influence autophagy of all substrates. For example, levels of α-synuclein do not change depending on serine-367 phosphorylation of PS1, he said. This could be due to the heterogeneity of lysosomes and vesicular compartments in neurons, as well as to the specific localization of protein substrates within those diverse vesicles, Greengard added. Even various products of the same substrate—APP—are not equally susceptible to autophagy. For example, following cleavage of APP by β-secretase, βCTF rapidly moves into lipid rafts, but any remaining full-length APP does not, Greengard said. This shift in localization could explain why βCTF, but not full-length APP, is fodder for autophagy, Greengard said.
Subcellular location also influences the activity of γ-secretase. For example, researchers led by Wim Annaert at KU Leuven in Belgium reported that unlike PS1, which occurs in various cell compartments, PS2 resides primarily in late endosomes, where it leans more toward production of Aβ42 than does PS1 does (see Jun 2016 news). Whether PS1’s phosphorylation at serine-367 affects its intracellular localization remains to be investigated.
Wolfe commented that follow-up work should address whether PS1 exerts its influence on autophagy when assembled with other members of the γ-secretase complex, or goes solo—essentially moonlighting in this separate function. Other researchers said they would like to see proof that serine-367 phosphorylation does not affect γ-secretase activity, as well as a detailed characterization of the vesicular compartments involved in βCTF degradation. Both Annexin A2 and Vamp8 are known to also associate with endosomes, for example, and endosomal markers were not included in the current study.
Why would PS1 have dual, apparently antagonistic roles? Greengard offered that both functions achieve a similar end: the destruction of βCTF. This could be beneficial, given reports that βCTF bungles endosomal trafficking independently of Aβ, he said (see Jul 2015 news). One recent study, led by Frédéric Checler at the University of Nice-Sophia-Antipolis in France, described how an accumulation of βCTF in neurons inhibited autophagy, kicking off a vicious cycle of malfunctions that derailed lysosomal trafficking (see Lauritzen et al., 2016). Checler commented that Greengard’s studies dovetail nicely with his findings.
How much PS1 is phosphorylated in the human brain? Does that change with age or disease? No one knows. Even so, the researchers proposed that increasing serine-367 phosphorylation could therapeutically reduce Aβ levels. They are conducting high throughput screens to find small molecules—either kinase activators or phosphatase inhibitors—to boost PS1 phosphorylation. Other parts of the autophagosome-lysosome pathway could serve as targets, as well, they told Alzforum.—Jessica Shugart
References
News Citations
- Perplexing Presenilins: New Evidence for Calcium Leak Channels
- Lodged in Late Endosomes, Presenilin 2 Churns Out Intraneuronal Aβ
- Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
Research Models Citations
Paper Citations
- Matz A, Halamoda-Kenzaoui B, Hamelin R, Mosser S, Alattia JR, Dimitrov M, Moniatte M, Fraering PC. Identification of new Presenilin-1 phosphosites: implication for γ-secretase activity and Aβ production. J Neurochem. 2015 May;133(3):409-21. Epub 2015 Feb 24 PubMed.
- Maesako M, Horlacher J, Zoltowska KM, Kastanenka KV, Kara E, Svirsky S, Keller LJ, Li X, Hyman BT, Bacskai BJ, Berezovska O. Pathogenic PS1 phosphorylation at Ser367. Elife. 2017 Jan 30;6 PubMed.
- Uemura K, Lill CM, Li X, Peters JA, Ivanov A, Fan Z, Destrooper B, Bacskai BJ, Hyman BT, Berezovska O. Allosteric modulation of PS1/gamma-secretase conformation correlates with amyloid beta(42/40) ratio. PLoS One. 2009;4(11):e7893. PubMed.
- Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, Hyman BT. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005 Mar 16;25(11):3009-17. PubMed.
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics. 2008 Sep;7(9):1598-608. Epub 2008 May 6 PubMed.
- Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016 Aug;132(2):257-76. Epub 2016 Apr 30 PubMed. Correction.
Further Reading
No Available Further Reading
Primary Papers
- Bustos V, Pulina MV, Kelahmetoglu Y, Sinha SC, Gorelick FS, Flajolet M, Greengard P. Bidirectional regulation of Aβ levels by Presenilin 1. Proc Natl Acad Sci U S A. 2017 Jul 3;114(27):7142-7147. Epub 2017 May 22 PubMed.
- Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, Greengard P. Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome-lysosome fusion. Proc Natl Acad Sci U S A. 2017 Jul 3;114(27):7148-7153. Epub 2017 May 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
KULeuven & VIB
Both these papers from the Greengard lab report on a bidirectional role of PS1 in APP processing— one through the known catalytic role in γ-secretase, and now an opposite role via the phosphorylation of the S367 residue, which is proposed to target βCTF to autophagosomal degradation, thereby reducing Aβ production. These findings support a general idea that processing and downstream signaling is finely balanced and that compensating mechanisms exist that prevent overproduction in healthy situations, which could be of therapeutic value.
Mechanistically, the authors provide evidence that PS1 interacts with AnnexinA2, which, through binding with VAMP8, facilitates interaction with the autophagosomal SNARE syntaxin17. These data support previous reports, including from the same lab, that part of the APP proteolytic regulation goes through the autophagy route.
Although appealing, I would remain cautious about such a mechanism, because it raises the question which pool of APP is subject to this. Generally, APP is processed at the surface and endosomal compartments, with early and late endosomes as major sites for Aβ production. Indeed, interfering with the ESCRT-dependent sorting of cargo, like APP, as shown by altering expression of vps34 (Morel et al. 2013) or CD2AP (Ubelmann et al. , 2017) the balance between amyloidogenic processing and sorting βCTF via ILV to lysosomal degradation. At least in this route, βCTF doesn’t need to encounter the autophagy route to reach lysosomes for degradation.
I am wondering whether the authors invoke a direct sequestration of a pool of APP to initiating autophagosomes, i.e., prior to fusion with the lysosome? This might be addressed by identifying the subcellular compartments where PS1 and AnnexinA2 are thought to interact. The proximity ligation assay (PLA) data as well as pull-down indicate that this interaction is abundant; therefore it should be feasible to combine this with organelle identification. The prediction of the authors is, given the role of AnnexinA2 in autophago-lysosomal fusion, that the PLA-positive spots should be on these related organelles.
On the other hand, it cannot be deduced from the data whether interaction occurs with the mature heterodimeric PS1 or with the full-length protein; in the latter case, the interaction should be confined to the ER-pool of PS1. This raises also another intriguing question: is this alternative function of PS1 mediated through the single protein, or through PS1 being associated within the γ-secretase complex? Further experimentation is required to characterize this interaction.
Of note, while here phosphorylation of S367 is protective, a recent publication showed that phosphorylation at this residue (and other residues) results in a more “closed” conformation of γ-secretase and thus an increase in Aβ42/40 ratios (Maesako et al., 2017).
References:
Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall'armi C, Simoes S, Point Du Jour KS, McCabe BD, Small SA, Di Paolo G. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4:2250. PubMed.
Ubelmann F, Burrinha T, Salavessa L, Gomes R, Ferreira C, Moreno N, Guimas Almeida C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017 Jan;18(1):102-122. Epub 2016 Nov 28 PubMed.
Maesako M, Horlacher J, Zoltowska KM, Kastanenka KV, Kara E, Svirsky S, Keller LJ, Li X, Hyman BT, Bacskai BJ, Berezovska O. Pathogenic PS1 phosphorylation at Ser367. Elife. 2017 Jan 30;6 PubMed.
The University of Tokyo
A γ-secretase-independent function of PS has been implicated in several signaling pathways (Duggan and McCarthy, 2016). Importantly, proteolytically inactive PS is biologically active in several organisms, including the moss P. patens (Khandelwal et al., 2007), in which the other γ-secretase components are not conserved. Thus, non-proteolytic function of PS in the membrane/protein trafficking might be one of the moonlighting roles of PS family proteins. This idea is also supported by biological properties of other pseudoproteases. iRhom proteins are catalytically inert rhomboids—the intramembrane cleaving serine proteases—and some of them regulate protein trafficking (Lemberg et al., 2016). In addition, proteolytically inactive DPP10 and ADAM11 regulate the subcellular localization of potassium channels (Nadal et al., 2003; Kole et al., 2015).
Although some results are controversial, this study and another recent paper Maesako et al. (2017) reminds us that the hydrophilic loop 6 is a functionally and pathologically important domain of PS1. The primary sequences of loop 6 of PS1 and PS2 are not conserved, and several specific binding partners have been reported, indicating that the hydrophilic loop 6 is involved in PS1/PS2-specific function. In addition, Deng et al. (2006) reported that genetic ablation of exon 10 of PS1, encoding amino acids 320-377, in mice increased the Aβ42 ratio and CTF levels in the brain. In addition, deletion of exon 9-10 identified in a patient with early onset AD exacerbated the pathological effect of exon 9 deletion in cultured cells (Le Guennec et al., 2017). Thus, further precise investigation of this region could provide novel therapeutic approaches to AD.
References:
Duggan SP, McCarthy JV. Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell Signal. 2016 Jan;28(1):1-11. Epub 2015 Oct 21 PubMed.
Khandelwal A, Chandu D, Roe CM, Kopan R, Quatrano RS. Moonlighting activity of presenilin in plants is independent of gamma-secretase and evolutionarily conserved. Proc Natl Acad Sci U S A. 2007 Aug 14;104(33):13337-42. PubMed.
Lemberg MK, Adrain C. Inactive rhomboid proteins: New mechanisms with implications in health and disease. Semin Cell Dev Biol. 2016 Dec;60:29-37. Epub 2016 Jul 1 PubMed.
Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003 Feb 6;37(3):449-61. PubMed.
Kole MJ, Qian J, Waase MP, Klassen TL, Chen TT, Augustine GJ, Noebels JL. Selective Loss of Presynaptic Potassium Channel Clusters at the Cerebellar Basket Cell Terminal Pinceau in Adam11 Mutants Reveals Their Role in Ephaptic Control of Purkinje Cell Firing. J Neurosci. 2015 Aug 12;35(32):11433-44. PubMed.
Maesako M, Horlacher J, Zoltowska KM, Kastanenka KV, Kara E, Svirsky S, Keller LJ, Li X, Hyman BT, Bacskai BJ, Berezovska O. Pathogenic PS1 phosphorylation at Ser367. Elife. 2017 Jan 30;6 PubMed.
Deng Y, Tarassishin L, Kallhoff V, Peethumnongsin E, Wu L, Li YM, Zheng H. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired gamma-secretase activity and exacerbated amyloid pathology. J Neurosci. 2006 Apr 5;26(14):3845-54. PubMed.
Le Guennec K, Veugelen S, Quenez O, Szaruga M, Rousseau S, Nicolas G, Wallon D, Fluchere F, Frébourg T, De Strooper B, Campion D, Chávez-Gutiérrez L, Rovelet-Lecrux A. Deletion of exons 9 and 10 of the Presenilin 1 gene in a patient with Early-onset Alzheimer Disease generates longer amyloid seeds. Neurobiol Dis. 2017 Aug;104:97-103. Epub 2017 Apr 28 PubMed.
Make a Comment
To make a comment you must login or register.