CRISPR Edits Genome of Human Embryos
Quick Links
Scientists have used the CRISPR-Cas9 precision gene-editing system to snip a disease-causing mutation right out of viable human embryos. They did so without accidentally cutting DNA elsewhere, or inducing a heretofore common problem with editing human DNA known as mosaicism, where embryos end up with a mix of edited and unedited cells. Further improvements will be needed before any embryos are implanted for pregnancy, but the research offers hope that this and some other autosomal-dominant mutations can be erased from future generations.
The August 3 Nature paper was led by an international team of researchers, including Paula Amato, Sanjiv Kaul, and Shoukhrat Mitalipov of Oregon Health and Science University, Portland, along with Juan Carlos Izpisua Belmonte from the Salk Institute for Biological Studies, La Jolla, California, and Jin-Soo Kim at the Institute for Basic Science, Daejeon, Republic of Korea. The news made headlines across the news media (see, e.g., The New York Times; The Atlantic; and Wired).

Dividing, Free of Mutation.
Two days after fertilization and injection with CRISPR-Cas9, early embryos grow without a disease-causing mutation. The embryos were not implanted. [Courtesy of OHSU.]
“This study is paving the way to CRISPR–Cas9 reaching the clinic in the future,” wrote Nerges Winblad and Fredrik Lanner at Karolinska University Hospital, Stockholm, in an accompanying News and Views. They added that although the technique is promising, further studies and optimization will be needed before it is considered safe for therapy. The authors readily agree.
“The study has direct implications for familial Alzheimer’s disease, but we are not ready for prime-time use of CRISPR in AD,” said Murali Doraiswamy, Duke University, Durham, North Carolina, who was not involved in the study. Other AD and amyotrophic lateral sclerosis (ALS) experts echoed the caution, saying the technique is promising for autosomal-dominant neurodegenerative disorders, but limited at the moment (see image above). For starters, the Alzforum mutations database lists about 320 different pathogenic mutations in APP, PS-1, PS-2, and tau known to cause dominantly inherited AD or other tauopathies, respectively. Scientists would have to target each one and study how well CRISPR repairs it. Pouring a bit more cold water on any excitement that may be heating up in the AD community, the authors themselves note that correcting point mutations, which make up a large majority of those that cause neurodegenerative diseases, is hard with the current technology.
To inform public debate about the ethics of editing germline DNA, the American Society of Human Genetics issued a position statement timed to the appearance of this paper.
Editing the Human Genome
Since its discovery in 2013, CRISPR-Cas9 has taken the research community by storm (Sep 2014 news series). Based on a bacterial defense system, this DNA-cutting technology directs a Cas9 enzyme to a particular spot on the genome using a matching guide RNA and creates a double-strand break there. The break is fixed either by non-homologous end joining (NHEJ), which introduces random insertions and deletions to turn off the gene, or by homology-directed repair (HDR), which uses a new piece of DNA—usually introduced along with CRISPR—as a template to insert a new sequence. CRISPR has been used three times before to edit germline DNA in human embryos (see Liang et al., 2015; Kang et al., 2016; Tang et al., 2017). Two of those studies used nonviable embryos; all three saw extensive mosaicism.
In the present study, first authors Hong Ma and Nuria Marti-Gutierrez at OHSU, Sang-Wook Park in Daejeon, and Jun Wu at the Salk wanted to see if CRISPR-Cas9 could correct a pathogenic mutation in viable human embryos without causing mosaicism. They reasoned that this could improve preimplantation genetic diagnosis (PGD), an extension of in vitro fertilization that doctors already use in rare cases to ensure that parents who carry dominant mutations have healthy children. With PDG, doctors test whether fertilized embryos have a genetic mutation, such as those for familial Alzheimer’s or Huntington’s disease, then implant only unaffected ones (July 2014 news series). What if affected embryos could be repaired to become implantable? That could reduce the number of rounds of in vitro fertilization cycles women have to endure before getting pregnant with an unaffected baby, Amato said at a press briefing.
To see if it was possible, the researchers chose to work on the gene encoding cardiac myosin-binding protein C. Mutations in MYBPC3 are a common cause of autosomal-dominant hypertrophic cardiomyopathy. In this disease, the heart muscles thicken, often unbeknownst to the victim, and can result in sudden death, most prominently in athletes. As in dominantly inherited AD and certain tauopathies, a single copy of the mutated allele suffices to cause disease.
A man with hypertrophic cardiomyopathy served as a donor in the experiments. The researchers derived induced pluripotent stem cells from his fibroblasts, used them to ascertain his exact mutation, then developed a guide RNA to match, and created a single-stranded oligodeoxynucleotide (ssODN) to serve as a corrected template for HDR. The CRISPR-Cas9 system they came up with cut DNA in 27 percent of the iPSCs grown in culture. About 40 percent of those were repaired by HDR and the ssODN, while NHEJ took care of the rest.
The researchers then used the patient’s sperm to fertilize eggs from 12 healthy egg donors, simultaneously injecting the CRISPR-Cas9 complex and the ssODN into half of the eggs. After three days, they tested each cell in every embryo to learn how many had two copies of the wild-type allele. About half of the untreated controls were homozygous for the wild-type allele, as would be expected in ordinary PGD. By contrast, 72 percent of the treated embryos had a double wild-type allele; this meant some of the affected ones had been repaired by HDR. The remaining 16 treated embryos showed signs of NHEJ, which is unhelpful for gene editing. Allowed to grow for five days, the embryos developed as they normally would.
Exploring the mechanism of repair yielded a surprise. These human embryos almost always used the wild-type strand on the healthy allele to guide the repair, rather than the introduced ssODN. In mice, it’s the opposite, where embryos more frequently use the ssODN as a template in HDR (Wu et al., 2013). The scientists could tell the difference because the ssODN included unique nucleotides that distinguished it from wild-type. This suggests humans and mice use different repair mechanisms in their embryos. The embryos’ mechanisms also appear to differ from iPSCs, which tended to use the ssODN for HDR.
Importantly, the researchers found that injecting the CRISPR-Cas9 complex at the same time as the sperm prevented mosaicism almost completely. They guessed that doing so ensured the gene editing would occur before the first division. By contrast, if CRISPR is injected even a short while after fertilization, it may operate after the zygote has already started dividing, correcting the mutation in only a subset of cells (see image below).
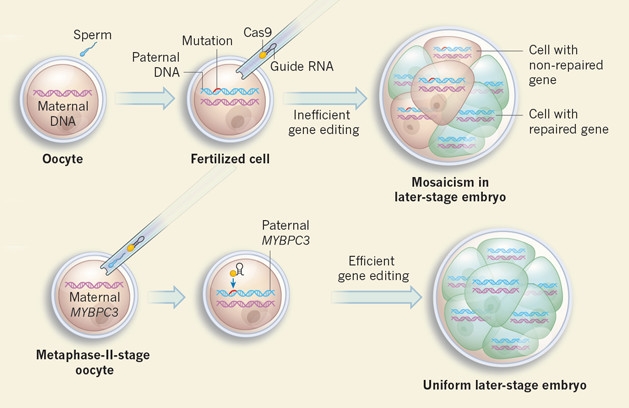
Mosaic Work-Around: CRISPR injected after fertilization operates after the first division and results in a subset of cells being fixed. Injecting CRISPR and sperm together ensures that repair occurs before the zygote has time to split. [Courtesy of Winblad and Lanner, Nature.]
Lastly, the authors scoured the rest of the embryos’ genes with whole-genome and exome sequencing, finding no evidence that the CRISPR complex had cut anywhere else. These off-target cleavages have been a big concern with CRISPR-Cas9.
Much work remains to be done before researchers can implant these embryos to result in pregnancy. For example, the technique needs to approach 100 percent efficiency, said Mitalipov. He plans to try small molecules that downregulate NHEJ and upregulate HDR, but needs to study whether embryos exposed to those compounds develop normally. Once such safety data is in hand, regulators will decide whether researchers can go ahead with clinical trials, Mitalipov said.
For now, the National Institutes of Health does not support research on gene editing in human embryos. Neither can the Food and Drug Administration consider clinical trials that deal with germline genetic modification. A 2017 report by the U.S National Academy of Sciences and National Academy of Medicine stipulated that germline gene editing should only happen in cases where there are no “reasonable alternatives,” such as PGD. Mitalipov hopes committees will loosen their restrictions once they see more evidence that the problems of mosaicism and off-target DNA changes have been solved.
Coinciding with Mitalipov’s paper, the American Society of Human Genetics released a statement saying that it is too early to perform germline editing that will result in pregnancy in people (Ormond et al., 2017). Ten other international organizations are part of the report, including the National Society of Genetic Counselors, the Human Genetics Society of Australasia, the Southern African Society for Human Genetics, and the Asia Pacific Society of Human Genetics. However, the group’s consensus supports germline genome editing research, with appropriate oversight and consent, that explores the relevant questions. Importantly, it supports public funding for such research.
What About Neurodegenerative Disease?
Will this technique work for other autosomal-dominant mutations? Eventually, yes, said Mitalipov, citing the breast and ovarian cancer mutations BRCA1 and 2 as examples. The specificity of CRISPR-Cas9 will depend on each individual mutation and the donor’s genetic background. Off-target effects likely will be more common for mutations that look similar to their wild-type alleles.
Kim cautioned that single nucleotide mutations are more challenging to correct than larger insertions or deletions such as the one targeted in this paper. Since the error in single base pair substitutions is so small in size, it will be difficult for Cas9 to home in on the mutant allele. This pertains in particular to neurodegeneration. “The report provides some clear translatability to many autosomal dominant Alzheimer’s disease (ADAD) mutations,” wrote Eric McDade, Washington University School of Medicine in St. Louis, to Alzforum. “However, the mutation that was the focus of this research was a deletion, and most ADAD-causing mutations are single base pair substitutions.” (See McDade’s full comment below.)
What about other neurodegenerative diseases? “This new success could be relevant for dominantly inherited ALS and FTD,” wrote Ronald Klein, LSU Health Sciences Center-Shreveport, to Alzforum. Between 10 and 20 percent of ALS and FTD cases are considered to be heritable, and most of the underlying mutations are dominant, Klein said. Of those, it might be promising to explore gene editing for C9ORF72 hexanucleotide repeats; also for ALS mutations in the genes NEK1, SOD1, TDP-43, FUS, and others; as well as FTD mutations in tau, progranulin, VCP, CHMP2B and other genes. As in Alzheimer’s, however, most ALS and FTD mutations are single nucleotide substitutions, Klein cautioned. Removing extra stretches of disease-causing repeats by CRISPR-Cas9 might work better, he agreed.
Not many ADAD families know about DNA repair with CRISPR just yet; however, McDade says he expects interest to grow in the near future, and Mitalipov’s paper is already being posted on private familial AD discussion groups. If the procedure becomes more efficient, less expensive, and is proven in models to lead to normal development, it will become more attractive, he said. Some families with ADAD mutations are using IVF/PGD already.
On the other hand, Ammar Al-Chalabi, King’s College London, who studies the human genetics of ALS, pointed out that PGD is simpler and still comes up with about half the embryos being healthy. Merit Cudkowicz, Massachusetts General Hospital, Boston, noted that if some of the allele-specific oligonucleotide (ASO) and gene therapies for ALS that are already going into people in clinical trials are effective, then families and clinicians may not need advanced PGD approaches employing CRISPR-Cas9.
“This paper is an important milestone in using CRISPR for genome editing of familial disease mutations in the germline,” said Martin Kampmann, University of California, San Francisco. Ahmet Yildiz, University of California at Berkeley, agreed. “This technique has great power to repair genetic diseases, and I believe we have to make the best use of it for health,” he said. Both Kampmann and Yildiz emphasized that ethical and safety standards have to be developed before this technology can be applied to patients.
For safety, limiting off-target cleavage by Cas9 will be critical. On this front, Yildiz, working with CRISPR-Cas9 co-discoverer Jennifer Doudna, also at Berkeley, on August 4 described in Science Advances why CRISPR-Cas9 cuts at specific target sequences in the genome, and where the tendency for off-target binding comes from. First authors Yavuz Dagdas and Janice Chen found that Cas9’s cutting region, the HNH domain, takes one conformation when it binds its guide RNA, and another when it cleaves DNA. It passes through a checkpoint intermediate to get from point A to point B, and has to break free of that intermediate before it can adopt its cleaving form. If CRISPR-Cas9 binds DNA with more than three mismatches to its guide RNA, the HNH domain cannot overcome this energy hump to cut. “Our work explains why Cas9 binds to many off-target sites but cleaves only a subset of them,” Yildiz wrote to Alzforum.
One way to improve that accuracy would be to adjust the guide RNA. While Cas9 with a guide RNA of 20 nucleotides tolerates several mismatches on the DNA, one with 17 is more sensitive to them, and so binds fewer off-target sequences. Scientists are also engineering Cas9 to make it more specific. “While these approaches significantly reduce off-target editing, none of them can fully eliminate cleavage of off-targets with a single mismatch at the moment,” Yildiz said. He agreed that variations of just a single base pair may not be trivial to edit with the current CRISPR technology.—Gwyneth Dickey Zakaib
References
Series Citations
Paper Citations
- Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, Lv J, Xie X, Chen Y, Li Y, Sun Y, Bai Y, Songyang Z, Ma W, Zhou C, Huang J. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015 May;6(5):363-372. Epub 2015 Apr 18 PubMed.
- Kang X, He W, Huang Y, Yu Q, Chen Y, Gao X, Sun X, Fan Y. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Genet. 2016 May;33(5):581-588. Epub 2016 Apr 6 PubMed.
- Tang L, Zeng Y, Du H, Gong M, Peng J, Zhang B, Lei M, Zhao F, Wang W, Li X, Liu J. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol Genet Genomics. 2017 Jun;292(3):525-533. Epub 2017 Mar 1 PubMed.
- Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013 Dec 5;13(6):659-62. PubMed.
- Ormond KE, Mortlock DP, Scholes DT, Bombard Y, Brody LC, Faucett WA, Garrison NA, Hercher L, Isasi R, Middleton A, Musunuru K, Shriner D, Virani A, Young CE. Human Germline Genome Editing. Am J Hum Genet. 2017 Aug 3;101(2):167-176. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. Application of the gene editing tool, CRISPR-Cas9, for treating neurodegenerative diseases. Neurochem Int. 2017 Jul 18; PubMed.
- Kampmann M. A CRISPR Approach to Neurodegenerative Diseases. Trends Mol Med. 2017 Jun;23(6):483-485. Epub 2017 May 4 PubMed.
Primary Papers
- Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park AR, Kim D, Kim ST, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JC, Amato P, Kim JS, Kaul S, Mitalipov S. Correction of a pathogenic gene mutation in human embryos. Nature. 2017 Aug 2; PubMed.
- Winblad N, Lanner F. Biotechnology: At the heart of gene edits in human embryos. Nature. 2017 Aug 2; PubMed.
- Ormond KE, Mortlock DP, Scholes DT, Bombard Y, Brody LC, Faucett WA, Garrison NA, Hercher L, Isasi R, Middleton A, Musunuru K, Shriner D, Virani A, Young CE. Human Germline Genome Editing. Am J Hum Genet. 2017 Aug 3;101(2):167-176. PubMed.
- Dagdas YS, Chen JS, Sternberg SH, Doudna JA, Yildiz A. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv. 2017 Aug;3(8):eaao0027. Epub 2017 Aug 4 PubMed.
- Egli D, Zuccaro MV, Kosicki M, Church GM, Bradley A, Jasin M. Inter-homologue repair in fertilized human eggs?. Nature. 2018 Aug;560(7717):E5-E7. Epub 2018 Aug 8 PubMed.
- Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, Beard MR, Hughes J, Pomerantz RT, Thomas PQ. Large deletions induced by Cas9 cleavage. Nature. 2018 Aug;560(7717):E8-E9. Epub 2018 Aug 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
The report provides an interesting approach using a heterozygous-dominant disease, and provides some clear translatability to many ADAD mutations. However, the mutation that was the focus of this research was a deletion, and most ADAD-causing mutations are single base pair substitutions.
It is not certain at this time how many people with known ADAD mutations in their families consider pre-implantation IVF as a way of having children while significantly decreasing the risk of passing on the genetic defect; however, this does not appear to be a common approach at this time. Therefore, I'm not certain of the impact DNA repair through CRISPR would have in the near future.
With that said, the current pre-implantation IVF screening approach is used by some with these mutations. The ability to make the procedure more efficient, if it has a cost impact, for instance, and is proven in the models used to lead to normal development, might make this approach more attractive.
The cost and potential medical record implications of undergoing current IVF/PGD likely limit its use in many of our families. However, during our recent Dominantly Inherited Alzheimer Disease Family Conference, held at University College London in July, there were ADAD family members who reported using pre-implantation IVF as part of their decision to have children, so it is certainly being used. This procedure, if safe, effective, and reproducible, might make this option more commonly used.
With these aspects in mind, I would certainly want to see this used in mutations with single nucleotide substitutions (variants) that characterize most ADAD mutations, and like to see the further development of the embryos as it relates to normal development after the normal sequence is introduced.
Of note, there are studies in Duchenne muscular dystrophy using CRISPR/Cas9 that are planned for Phase 1 in the near future. It is certain that the researchers and families with familial dementias will be following these studies closely.
Make a Comment
To make a comment you must login or register.