Consortium Debuts Biggest Set of Human Epigenomes To Date
Quick Links
A consortium of 258 researchers at 41 institutions, having completed 2,800 experiments and sequenced 150 billion DNA fragments over seven years, now presents the human epigenome. In the February 19 Nature, corresponding author Manolis Kellis of MIT and colleagues in the Roadmap Epigenomics Consortium describe 111 epigenomes from various tissues in healthy adults, fetuses, and cell lines. The epigenomes will serve as reference data. The scientists profiled eight different regions of the brain, including the hippocampus, substantia nigra, and prefrontal cortex. The data are freely accessible online.
The reference epigenomes profile normal tissues, but in another paper in the same issue of Nature, Kellis and his colleague Li-Huei Tsai describe an Alzheimer’s-related epigenome. They compared the hippocampal epigenome of a mouse AD model to genetic hits from genome-wide association studies (GWAS). They report that many Alzheimer’s-linked mutations tend to affect not neural activities such as learning and memory, but immune-system functions. The immune system has stood out in other studies of AD gene expression and in network analyses as well (see Jan 2015 news; May 2013 Webinar). Changes to gene expression in immune cells might be at the root of Alzheimer’s disease, Kellis suggested.
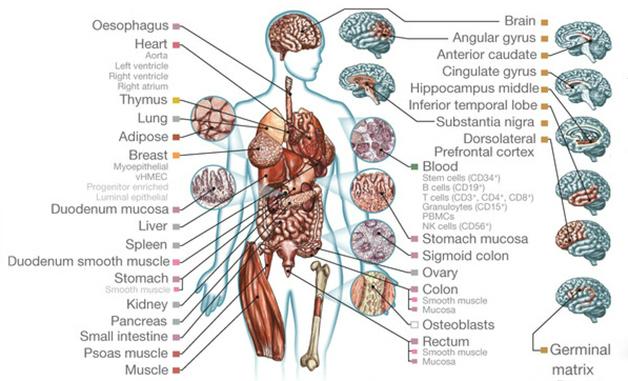
Epigenome Laid Bare. Researchers deciphered the epigenomes of 111 different tissues, including eight regions of the brain. [Image courtesy of Roadmap Epigenomics Consortium et al., Nature.]
Funded by the National Institutes of Health Common Fund, the Roadmap project was initiated in 2008 in the wake of the first human genome sequence, which was completed in 2003. The genome’s As, Ts, Gs, and Cs delineate the basic set of instructions to make a person, but the epigenome determines which instructions each cell type will enact, Kellis said. The epigenome takes the form of a variety of decorations on the DNA, as well as the histone protein drums that spool the nucleic acid strands. Both DNA and histones can pick up methyl groups, and histones can be acetylated as well. Depending on exactly where they occur, these modifications help determine whether a gene is read or ignored by the transcription apparatus. Another feature of the epigenome is how tightly the DNA encircles those histones. Loosely wound DNA is more accessible to RNA polymerase, and thus more likely to be transcribed.
The Roadmap effort builds on the Encyclopedia of DNA Elements (ENCODE) project, which identified regulator regions in the genome (see Sep 2012 news; Sep 2014 news). However, where ENCODE focused on genome annotation in a handful of cell types, the Roadmap zeroed in on epigenetic marks in a much larger set of tissues.
In one of the Nature papers, joint first authors Anshul Kundaje, Wouter Meuleman, and Jason Ernst of MIT, and Misha Bilenky of the BC Cancer Agency in Vancouver, British Columbia, Canada—along with their 200-plus collaborators—lay out what they describe as “the largest collection so far of human epigenomes.” Ammar Al-Chalabi of King’s College London, who was not involved in the project, called it an essential resource (see full comment below).
Curating the Epigenome
The Roadmap researchers initially set a goal of just 20 epigenomes, but rapid improvements to DNA sequencing allowed them to expand their plans, Kellis said. Collaborators at hospitals collected samples from normal adults and fetuses, including tissues from heart, stomach, skin, fat, and blood. “Every sample has a different story,” said Kellis. The goal was to create reference epigenomes for healthy tissues first; profiling of diseased tissue will happen in the future. However, even the normal epigenomes can yield insight into disease, because they help scientists figure out how disease-related genes are regulated in different tissues.
Included were brain samples pooled from two people who participated in the ongoing Memory and Aging Project at the Rush Alzheimer’s Disease Center in Chicago. One was a man who had died at the age of 81, the other a woman who had died at 75, said Philip De Jager of Brigham and Women’s Hospital in Boston, who co-selected the samples with David Bennett at Rush. Both people were cognitively sharp until death, and exhibited minimal brain pathology on autopsy. “We tried to find the cleanest brains that we could,” so the epigenome would reflect a healthy brain, De Jager said.
The consortium also analyzed a handful of embryonic stem cell and induced pluripotent stem cell lines, and early stage derivatives of those embryonic stem cells such as neuronal progenitors. They analyzed epigenome changes and transcription-factor binding during stem cell differentiation into neurons and other cell types in three additional papers in the February 19 Nature. Understanding the epigenomes of different cell types could help scientists figure out which regulators could be used for reprogramming iPS cells to their desired cell type, Kellis said.
There are many kinds of epigenetic marks, and it takes a different experiment to pick up each one. “It would be impossible in this day and age to complete an entire epigenome,” Kellis pointed out. “We do not even know all the histone modification marks that exist.” Therefore, the researchers selected a set of five “core” epigenetic marks to identify in all 111 samples. All five are methylations of the histone H3; however, they have different functions. For example, a triplet of methyls on the lysine at position 36 often indicates that the DNA wrapped around that histone is actively transcribed, while trimethylation on the lysine at position 27 suggests repression. To identify where in the genome such modified H3s are bound, the researchers used antibodies to each specific modification. They pulled down bits of DNA that bound those antibodies, and sequenced the fragments.
The authors analyzed those five core histone markers for all 111 tissues they examined. For a subset of tissues, they looked at other epigenetic markers as well. For example, they used the same antibody technique to examine histone acetylation or DNA methylation. They also measured the DNA’s sensitivity to DNase, which indicates that the DNA is unwound and active. This work generated 111 data sets that have at least the core histone methylations, and some data sets have more.
These publicly available epigenomes are currently in first-draft form. Every data set includes a set of quality measures, such as how many times a bit of DNA was read, or the signal-to-background ratio, so users get a sense of how robust the data is. “[The data] are all good, though some are much higher quality than others,” Kellis said. Ekaterina Rogaeva of the University of Toronto, who was not involved in the project, noted she would replicate any findings for her favorite genes in her own lab, just to be sure the epigenetic markers are there.
The Roadmap project was intended to be a one-time endeavor. It has come to an end, but other groups have already started the next steps. The International Human Epigenome Consortium (IHEC) aims to map 1,000 epigenomes, from both human and model organism tissues. Similarly, human genome-sequencing efforts grew from that first one to 1,000 genomes and more (see Nov 2012 news). Meanwhile, the Genotype-Tissue Expression (GTEx) project will address how much the epigenome might vary from person to person. GTEx investigators will profile multiple tissues across hundreds of individuals, Kellis said. The Roadmap, in contrast, offers just one epigenome per tissue type, from one or a pool of tissue donors, much like the original Human Genome Project did.
One shortcoming of the data set is that researchers profiled a given tissue using whole populations of cells, and hence would have missed cell-to-cell variations, note Casey Romanoski and Christopher Glass of the University of California, San Diego, in a commentary accompanying the Nature papers. The tissues include diverse cell types. De Jager pointed out that the brain samples, while mostly neurons and glia, contain other types like endothelial cells and pericytes too. Kellis said the cell lines have the benefit of being pure monocultures, whereas the tissues offer a better representation of what happens in a human body.
As more detailed epigenomes continue to emerge, what can researchers studying neurodegeneration do with the information? Kellis and colleagues offer a taste of the possibilities with an analysis of several diseases, including Alzheimer’s. GWAS have identified genetic loci where variants affect disease risk, but these are often in non-coding regions and the field has struggled to determine how such variants affect biology. Many variants are located in enhancer regions that may be kilobases away from the gene(s) they control.
The reference epigenomes can help interpret those GWAS hits, the Roadmap collaborators explained. For example, the epigenomes show in which cell types these variants are most active. The researchers compared GWAS variants from 58 different diseases with active regions of the genome as seen on the epigenome maps of different tissues. GWAS hits linked to high blood pressure, for instance, tended to occur in or near enhancers active in the heart, while variants associated with immune disorders, such as multiple sclerosis, mapped to enhancers utilized by immune tissues, such as T cells.
That may seem like no big surprise, Kellis conceded, but screening the epigenomes in this way provides an unbiased way to correlate GWAS hits to tissue types. It also turned up something unexpected. “Alzheimer’s was perhaps the biggest surprise,” he said. The genetic variants from GWAS mostly mapped to enhancers active in immune cells, not brain tissue.
Alzheimer’s, a Disease of the Innate Immune System?
That finding might have led Kellis to recheck his methods, he said, except he already had been working with Tsai on the AD epigenome study. It was also published in Nature and implicated the immune system. Co-first authors Elizabeta Gjoneska and Andreas Pfenning analyzed the epigenome of a model for AD, the CK-p25 mouse. In this inducible model, p25, a regulator of the brain-expressed, pro-apoptosis cyclin-dependent kinase 5 (Cdk5), can be overexpressed in the forebrain (Cruz et al., 2003). Cdk5 activity and p25 expression have been reported to be upregulated in the brains of people with Alzheimer’s and Parkinson’s disease (reviewed in Camins et al., 2006). The mouse exhibits several characteristics of Alzheimer’s, including excess amyloid peptide, tau aggregation, and neuron loss (Fischer et al., 2005; Cruz et al., 2006).
Gjoneska and Pfenning examined seven different kinds of histone modification in the hippocampi of these mice and in their uninduced littermates. Based on these markers, they inferred the location of 57,840 promoters and 151,447 enhancers. Of these, 5,056 promoters and 2,154 enhancers were less active, based on their histone markers, in the induced mice than in the controls. These sites were primarily regulators of genes for neural functions like learning and memory, as one might expect for a neurodegenerative condition. In addition, 3,667 promoters and 2,456 enhancers were more active in the induced mice. These were in regions known to influence expression of immune genes.
By comparing their data to the Roadmap human epigenomes, Tsai and colleagues were able to find the orthologs to many of these promoters and enhancers in the human genome. Then, they compared the pattern of up- and downregulated genetic elements from the CK-p25 mice to hits from Alzheimer’s GWAS (see Oct 2013 news). The AD genetic variants often landed within the upregulated, immune-system enhancers, but not in the downregulated, neural ones.
Kellis interpreted the results to mean that AD risk variants alter gene expression in immune cells, and suggested those changes could predispose someone to the disease. The changes to gene expression in the AD brain, in contrast, could be the result of environmental factors—education, for example—or consequences of the immune activation, Tsai said.
Tsai’s findings add to an emerging body of work that is pointing to the innate immune system’s contribution to Alzheimer’s, said De Jager, who was not involved with the AD mouse paper (reviewed in Heneka et al., 2015). De Jager also observed that certain AD GWAS hits regulate gene expression in monocytes, a type of immune cell (see May 2014 news). The immune system probably contributes to AD susceptibility and reacts to the pathology once it starts; the exact details are an open question, De Jager said.
Many genes that were upregulated in the CK-p25 mouse shared a binding motif for the transcription factor PU.1, which is highly expressed in monocytes and microglia. Tsai thinks PU.1 acts as a “master regulator” that turns up many immune response genes (see Feb 2015 conference news). If so, Kellis speculated, a therapeutic that deactivates PU.1 might be able to affect all those immune genes, and thus create a neuroprotective gene-expression profile. To test this idea, Tsai’s group plans to cross mouse models of neurodegenerative disease with PU.1 knockout mice.
Both De Jager and Rogaeva said they would be interested to see the epigenomic profile from a more frequently used mouse model of AD, with mutations in tau or APP. Tsai replied that her model captures AD pathology well, and that its gene-expression profile, based on RNA sequencing, lined up nicely with the RNA profile of the brains of people who had Alzheimer’s. The correlation between AD GWAS hits and immune genes upregulated in the CK-p25 mouse does suggest the model can offer useful information, said Rogaeva. Epigenetic profiles could help researchers understand which mouse best matches the human AD epigenome, De Jager suggested.
Rogaeva also pointed out that the immune system has been implicated in neurodegenerative diseases besides AD, including amyotrophic lateral sclerosis and Parkinson’s (see Dec 2011 news; May 2011 news). Might those diseases have the same immune cell epigenome signature as Alzheimer’s, she asked, or a different one? These kinds of studies might suggest therapeutic targets, Rogaeva said, just as the AD work pointed to PU.1.—Amber Dance
References
News Citations
- Network Analysis Points to Distinct Effects of Amyloid, Tau
- ENCODE Turns Human Genome From Sequence to Machine
- ENCODE Expands Analysis of DNA Regulation
- Genetics Project Update: Over 1,000 Genomes and Counting
- Paper Alert: New Alzheimer’s Genes Published
- Alzheimer's GWAS Hits Reflected in Monocyte Gene Expression
- Nature Versus Nurture: What Gives Microglia Their Identity?
- TDP-43 Hangs Out With NF-κB, Puts Innate Immunity Into Hyperdrive
- Interferon-γ’s Interference May Cause Parkinsonism
Webinar Citations
Paper Citations
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003 Oct 30;40(3):471-83. PubMed.
- Camins A, Verdaguer E, Folch J, Canudas AM, Pallàs M. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect. 2006 Oct;19(8):453-60. PubMed.
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005 Dec 8;48(5):825-38. PubMed.
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006 Oct 11;26(41):10536-41. PubMed.
- Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015 Mar;16(3):229-36. PubMed.
External Citations
Further Reading
Papers
- Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P, Polke JM, Sweeney MG, Mudanohwo E, Nacmias B, Sorbi S, Tartaglia MC, Rainero I, Rubino E, Pinessi L, Galimberti D, Surace EI, McGoldrick P, McKeever P, Moreno D, Sato C, Liang Y, Keith J, Zinman L, Robertson J, Rogaeva E. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015 May;129(5):715-27. Epub 2015 Feb 26 PubMed.
- Lunnon K, Mill J. Epigenetic studies in alzheimer's disease: Current findings, caveats, and considerations for future studies. Am J Med Genet B Neuropsychiatr Genet. 2013 Sep 13; PubMed.
- Devall M, Mill J, Lunnon K. The mitochondrial epigenome: a role in Alzheimer's disease?. Epigenomics. 2014;6(6):665-75. PubMed.
- Zhao YQ, Jordan IK, Lunyak VV. Epigenetics components of aging in the central nervous system. Neurotherapeutics. 2013 Oct;10(4):647-63. PubMed.
- Belzil VV, Bauer PO, Gendron TF, Murray ME, Dickson D, Petrucelli L. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 2014 Feb 12; PubMed.
News
- Newly Mapped DNA Elements Help Interpret GWAS
- Alzheimer’s Brains Mottled with Epigenetic Changes
- Epigenetic Alterations Mark Alzheimer’s Disease Genes
- Nature Versus Nurture: What Gives Microglia Their Identity?
- Newly Mapped DNA Elements Help Interpret GWAS
- Bethesda: "Ome" Sweet "Ome"—Epigenome Joins Genome, Proteome
- Methylation a Turn Off for Disease Gene C9ORF72?
- Alzheimer's GWAS Hits Reflected in Monocyte Gene Expression
Primary Papers
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature. 2015 Feb 19;518(7539):317-30. PubMed.
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015 Feb 19;518(7539):365-9. PubMed.
- Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. Epigenomics: Roadmap for regulation. Nature. 2015 Feb 19;518(7539):314-6. PubMed.
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015 Feb 19;518(7539):331-6. PubMed.
- Tsankov AM, Gu H, Akopian V, Ziller MJ, Donaghey J, Amit I, Gnirke A, Meissner A. Transcription factor binding dynamics during human ES cell differentiation. Nature. 2015 Feb 19;518(7539):344-9. PubMed.
- Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, Issner R, Gifford CA, Goren A, Xing J, Gu H, Cacchiarelli D, Tsankov AM, Epstein C, Rinn JL, Mikkelsen TS, Kohlbacher O, Gnirke A, Bernstein BE, Elkabetz Y, Meissner A. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature. 2015 Feb 19;518(7539):355-9. Epub 2014 Dec 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Kings College London
The Roadmap Epigenomics Consortium has set out an essential resource for researchers studying the epigenome. There are a number of key findings, among which are that epigenomic data sets can be imputed at high resolution and that disease-associated genetic variants show epigenomic enrichments in tissues relevant to the disease. This also means that where enrichment is seen in a tissue, it can be regarded as important for that disease. For Alzheimer’s, researchers find that immune cells show a signal consistent with the genetic evidence that the immune response plays a part in Alzheimer’s disease. An interesting aspect of this finding is that regions of the genome that do not quite reach genome-wide significance in GWAS are implicated by epigenomics, providing additional support for their involvement. In other words, the genomic and epigenomic evidence support each other.
View all comments by Ammar Al-ChalabiCardiff University
Epigenetics is an exciting and developing field. These papers represent an excellent first step to allow researchers to unravel the epigenetic architecture of diseases such as Alzheimer’s. The epigenomic roadmap provides a platform to advance our understanding of the genetic associations and biological pathways identified as important in disease etiology. The work on animal models further supports the primary role of immunity in disease, which has been highlighted by the function of a number of the AD susceptibility genes, and pathway analyses of genome-wide association datasets. It will be of great interest to the Alzheimer’s field to see how these findings from animal models translate into human studies.
University of Toronto
The wealth of data laid out by the Roadmap Epigenomics Consortium is both exciting and overwhelming. By adding together several layers of data, including multiple measures of DNA methylation, histone methylation marks, DNaseI digestion, and RNA sequencing, we can begin to build a more complete picture of how gene transcription is regulated in various cell types. Comparing pluripotent, multipotent, and fully differentiated cells gives us a glimpse into how epigenetics plays a role in lineage specification and helps us determine which genes are vital for this process.
Environmental contributions to complex diseases like Alzheimer’s have always been hard to define. We are now beginning to understand how things such as diet and exercise can influence gene transcription by altering their epigenetic regulation. The data presented by the Consortium will help us narrow our focus in this regard. By defining transcriptionally active/repressed genes in the brain and understanding the epigenetic signature responsible for that state, we can start selecting candidate genes whose transcription could be altered by the environment. The caveat is that people are not a homogeneous population of lab animals. Our challenge for the future will be to determine how variable these epigenetic signatures are across populations.
Make a Comment
To make a comment you must login or register.