To Clot...or Not? Aβ Promotes Fibrin Thrombosis in AD Mouse Models
Quick Links
The relationship between vascular risk factors and Alzheimer disease pathology has been debated for many years (see ARF Live Discussion and de la Torre, 2010). However, direct connections between AD-specific processes and cerebrovascular disease have remained unclear. In the June 10 issue of Neuron, a research team led by Sidney Strickland at Rockefeller University in New York City provides a link between the β amyloid peptide (Aβ) and abnormal fibrin clot formation in cerebral blood vessels. The authors propose that association between fibrin and Aβ may contribute to AD pathology and cognitive decline. In essence, they suggest a new role for Aβ in AD, whereby it would accumulate in the brain with aging and associate with fibrinogen. Given the pro-thrombotic environment present in AD, Aβ would lead to persistent clots that obstruct blood flow and engender inflammation.
“There is a lot of evidence that Aβ and circulatory problems are both involved in AD,” said Strickland in an interview with ARF. “However, there was previously no known mechanistic connection between the two.”
Several studies have linked increased fibrinogen and Aβ deposition with neurovascular damage and AD (see van Oijen et al., 2005; Paul et al., 2007; Smith and Greenberg, 2009). In this report, first authors Marta Cortes-Canteli and Justin Paul further investigated the relationship between fibrin and AD pathology by examining fibrin clot formation and degradation in the presence of Aβ protein in vitro and in vivo.
The authors first saw that purified fibrin clots formed in the presence of Aβ were slower to dissolve than clots formed without Aβ. Besides lingering longer, these clots were also structurally abnormal and, unlike normal clots, tended to cluster into irregular aggregates. When the researchers watched fibrin clot lysis under a microscope, they again saw delayed degradation of clots formed in the presence of Aβ. They often observed that fibrin/Aβ aggregates remained even after the surrounding fibrin network was degraded.
Strickland’s team then took these observations in vivo by examining fibrin clot dynamics in the TgCRND8 AD transgenic mouse model. By creating an imaging window in the skulls of living mice, the researchers were able to monitor the formation and lysis of clots in real time. Cortes-Canteli and colleagues induced clot formation either broadly by applying increasing concentrations of the damaging compound iron chloride ¬¬to the brain surface, or topically in individual blood vessels by using a two-photon laser scanning microscope. They saw that TgCRND8 mice formed fibrin clots at lower iron chloride concentrations than wild-type mice, and that their blood vessels became more blocked following laser-induced thrombosis. In addition, vessels containing vascular amyloid deposits, or cerebral amyloid angiopathy (CAA), tended to show more complete blockage than vessels lacking CAA. As in the in vitro experiments, blood clots in TgCRND8 animals resisted degradation. Furthermore, even before the scientists stimulated clotting, TgCRND8 mice tended to form blood clots spontaneously, unlike wild-type mice.
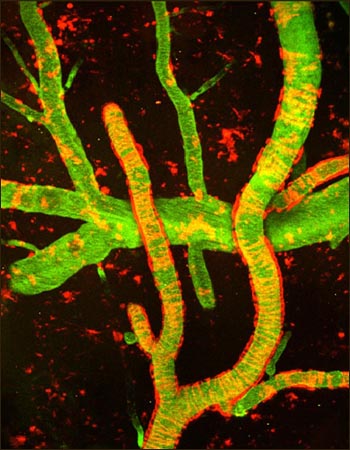
Multiphoton image of cerebral amyloid angiopathy (red) on blood vessels (green) in AD mouse model. On Neuron cover with the clever title, The Clot Thickens. Image credit: Marta Cortes-Canteli et al. Neuron. 2010 June 10
What might be the functional relationship between fibrinogen and Aβ? To address this question, the Rockefeller scientists examined the brain tissue of TgCRND8 mice for clues as to whether fibrinogen and Aβ interact within cerebral blood vessels. They saw that fibrinogen deposits often closely co-localized with CAA deposits, both in and around blood vessel walls. They also saw a similar association between fibrinogen deposit- and CAA-containing blood vessels in the postmortem brain tissue of AD patients.
This association between fibrinogen and vascular amyloid led the team to examine whether decreasing fibrinogen levels would also reduce CAA pathology. This time, they performed their experiments using two different AD mouse models, TgCRND8 and Tg6799, Robert Vassar’s 5x transgenic line. Researchers reduced fibrinogen blood levels by treating mice for four weeks with therapeutic doses of ancrod, a serine protease from a venomous viper. They then examined CAA pathology in the mouse brains. Ancrod treatment approximately halved plasma fibrinogen levels; it also reduced vascular amyloid within cerebral blood vessels by about a third. Similar reductions in CAA levels occurred in TgCRND8 mice in which fibrinogen levels were reduced by expressing a deficient copy of the fibrinogen gene, the scientists report.
However, as first author Cortes-Canteli said, “the final thing that everyone wants to know when you are talking about AD is whether cognitive impairment is decreased or prevented.”
The team testing the learning and spatial memory of TgCRND8 mice in the Morris water maze. TgCRND8 mice normally showed significant memory impairment compared with wild-type mice. However, TgCRND8 mice treated with ancrod improved to where their memory became comparable to wild-type performance. TgCRND8 mice expressing a deficient copy of the fibrinogen gene similarly improved their memory following ancrod treatment in a novel-arm Y maze memory task.
The authors indicate that these improvements in memory and CAA pathology seem to be due solely to decreased fibrinogen levels, as the total amount of Aβ protein in mouse brains was unchanged following ancrod treatment. “This could potentially be useful [therapeutically],” explained Strickland, “because the mechanism we propose is a specialized interaction between the Aβ peptide and the fibrin clot. If you could block that clotting interaction, you could theoretically block some of the deleterious consequences associated with AD without affecting general clotting where Aβ isn’t present.”
In interviews with ARF about this study, Jack de la Torre from the Banner Sun Health Research Institute, Sun City, Arizona, and Steven Greenberg from Massachusetts General Hospital in Boston both cautioned that observations made in mouse models can be difficult to extend to human disease and may not lead directly to viable treatment options. Still, both scientists, who were not involved in this study, said that this paper nicely establishes a link between fibrin clot formation and Aβ protein that warrants further investigation. In future experiments, de la Torre would like to know whether decreasing fibrinogen levels has a measurable effect on rates of cerebral blood flow, as this might more closely correlate the learning deficits seen in mice with the vascular risk factors seen in human patients.
“Ultimately, you want something that will help people with decline in cognitive function,” noted Strickland. “If you can do that, especially in a disease that is projected to be so catastrophic in an aging population, that would be incredibly satisfying.”—Elizabeth Eyler
Elizabeth Eyler is a freelance writer in Baltimore, Maryland.
References
Paper Citations
- de la Torre JC. Vascular risk factor detection and control may prevent Alzheimer's disease. Ageing Res Rev. 2010 Jul;9(3):218-25. PubMed.
- van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005 Dec 1;36(12):2637-41. PubMed.
- Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med. 2007 Aug 6;204(8):1999-2008. PubMed.
- Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009 Jul;40(7):2601-6. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010 Jun 10;66(5):695-709. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
NYU Medical Center
Regarding the true primary cause of Alzheimer's, the full pathway must be elucidated from start to finish. But it's important to consider that not every etiologic mechanism is therapeutically tractable. Sometimes it is better to approach a disease pathway using points downstream. Dysregulation of fibrinogen, amyloid-β, or some other molecule, may prove to be the initial driving force in this disease, but impaired blood flow could still be the best therapeutic target to improve cognition.
We already know that impaired blood flow can affect cognition. Alzheimer's may present a very subtle form of this, but we must keep our attention on the basic goal of therapy—to improve brain function. In medical school, I saw many patients diagnosed with Alzheimer's, and none of them first came to the physician complaining of plaques and tangles. They complained of problems with memory and cognition. We must always consider this when making strides in translational, "bench-to-bedside" research.
Make a Comment
To make a comment you must login or register.