ApoE4 Makes All Things Tau Worse, From Beginning to End
Quick Links
Today in the journal Nature, a large research collaboration led by David Holtzman of Washington University, St. Louis, formally published results previously presented at this year’s AD/PD conference (April 2017 conference news). The paper chronicles how ApoE interacts with tau to exacerbate, step by step, the pathogenic cascade underlying neurodegenerative disease, and does so independently of Aβ. The results cast the lipoprotein in an even darker light than before. They suggest that, far beyond its role as the strongest genetic risk factor for late-onset Alzheimer’s, ApoE may in fact fuel the entire neurodegenerative process across multiple diseases. “I think the work suggests we should now look at the role of ApoE in other tauopathies and other neurodegenerative diseases, not on risk to get the disease, but whether ApoE isoforms/ApoE influence progression,” Holtzman wrote to Alzforum (see Q&A below). The work also raises the profile of ApoE as a drug target.
- Deleting ApoE from FTD tau mice protects them against neurodegeneration.
- ApoE, especially E4, increases tau accumulation, redistribution to neuronal cell bodies, neuroinflammation, and brain atrophy.
- In human non-Aβ tauopathies, ApoE4 carriers have worse neurodegeneration; in AD, ApoE4 carriers progress faster.
As reported at AD/PD, first author Yang Shi and colleagues generated transgenic mice overexpressing the frontotemporal dementia tau mutation P301S on a background of either human ApoE2, 3, or 4, or no ApoE at all. Strikingly, the absence of ApoE strongly protected these tangle-bearing mice from developing the neurodegeneration and brain atrophy for which they are known. On the other hand, mice expressing the human ApoE4 isoform along with the mutant tau fared worst on all measures. In essence, this suggests that ApoE plays a part in neurodegeneration from soluble tau accumulation to brain atrophy, and that the specific mechanisms by which tau and ApoE interact likely change as pathogenesis marches on over time.
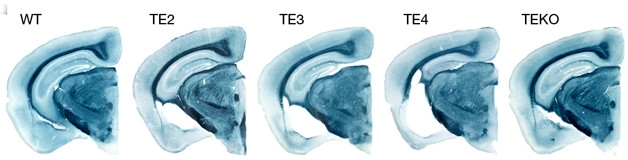
ApoE Shrank the Brain. In these slices, the hippocampal formation is at full size in wild-type (WT), nearly full size in FTD tau mice without ApoE (TEKO), and increasingly atrophied in FTD tau mice expressing ApoE2 (TE2), ApoE3 (TE3), or ApoE4 (TE4). [Courtesy of David Holtzman.]
At three months of age, before tau tangles form, P301S tau/ApoE4 mice had more soluble tau in the brain, and more phospho-tau staining in the hippocampus, than mice of the other ApoE genotypes. This was not due to production of tau. Rather, differences in expression of autophagy genes suggested that the E4 mice cleared tau more slowly, confirming a prior report in which Daniel Michaelson and colleagues had linked ApoE to impaired autophagy (Simonovitch et al., 2016).
By nine months of age, the excess tau in ApoE4 mice showed up in insoluble fractions when measured biochemically. Histologically in the hippocampus, p-tau was redistributed from axons to cell bodies in the tau/E4 mice. By this age, distinctive p-tau staining patterns emerged, whose increasing intensity going from ApoE 2 to 3 to 4 correlated with increasing atrophy. This suggests that ApoE affects something about tau’s conformation, or the progression of its pathology, the authors write.
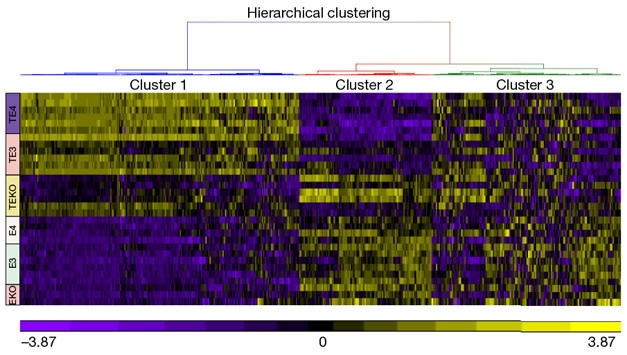
The Heat Is On. In this heatmap of microglial gene expression, pro-inflammatory genes (cluster 1, top left), are up the most, and homeostatic genes (cluster 2) down the most, in mice expressing FTD tau and ApoE4 (purple TE4, top rows). Mice expressing FTD tau but no ApoE (yellow TEKO rows) are protected. [Courtesy of David Holtzman.]
The presence of degenerating neurons induces neuroinflammation and here, too, ApoE made a difference. Shi et al. first confirmed a prior finding by Carol Colton and others that ApoE4 comes with higher innate immune reactivity than E2 or 3 do; then they profiled microglial gene expression. As presented at AD/PD, pro-inflammatory genes were up- and homeostatic genes downregulated in microglia from the tau/E4 mice. The paper shows that this profile became more pronounced along with the ApoE isoform-dependent p-tau staining pattern. This type of microglial activation did not occur in mice expressing only mutant human tau or only ApoE—or, for that matter, in three-month-old mice expressing both P301S tau and human ApoE. This means tau pathology must already be in place for microglia to rev up in this ApoE-dependent way.

ApoE4 Activates Astros.
A1-astrocyte-specific genes are up in neurons from mice expressing both FTD tau and ApoE4 (TE4 9 months, middle quadrant). This happens after neurodegeneration has started (TE4 3-months). [Courtesy of David Holtzman.]
Similarly, the finding, reported at AD/PD, that A1 astrocytes were strongly upregulated in these P301S tau/ApoE4 mice was not seen until the mice were nine months old. This suggests, again, that this neurotoxic switch occurs as a result of neurofibrillary pathology activating pro-inflammatory microglia, which then induce A1 astrocytes. Looking at brain tissue, the authors report that the astrocyte marker GFAP correlated tightly with brain atrophy, implying that the reactive astrocytes may be killing off neurons.
Not presented at AD/PD were experiments recapitulating the ApoE effects in vitro. The paper reports that P301S tau neurons died in droves when co-cultured with glia from ApoE4 knock-in, but not from ApoE knock-out mice. Abundant TNF-α floated in the medium from the former co-cultures, but not the latter. Finally, simply adding recombinant ApoE, especially the E4 variety, to cultured P301S neurons shrank the neurons’ arbors and caused some to die. This means a fraction of the overall ApoE effect could be due to a direct toxicity to neurons rendered vulnerable by tau pathology. That said, the neuron death in this assay was less extensive than in the neuron-glia cocultures, suggesting that neuroinflammation fans the flames of whatever damage tau and ApoE wreak on their own.

ApoE carriers with mild AD progress faster than non-carriers.
Do these data translate to humans? In a neuropathology series from 79 people who had died with corticobasal degeneration, Pick’s disease, and progressive supranuclear palsy, ApoE4 carriers had more severe regional neurodegeneration, the authors report. ApoE4 also appeared to speed progression of Alzheimer’s disease, which features both amyloid and tau pathology. In 592 people with initially mild symptomatic AD as confirmed by low CSF Aβ who were observed for 10 years, dementia worsened 23 percent faster in ApoE4 homozygote and 14 percent faster in ApoE4 heterozygote people than in E4 noncarriers. This meant that with or without Aβ pathology present as well, ApoE4 hastens the neurodegeneration that arises from tauopathy.
In toto, this study suggests that ApoE is detrimental along the cascade from protein aggregation to neuronal death. ApoE acts directly on tau-laden neurons, as well as indirectly via glia once the presence of degenerating neurons has provoked those cells. ApoE4 is always the worst.
This toxic gain of function might clarify how drug developers view the lipoprotein, which since its discovery as an AD risk factor in 1993 has not been the subject of a single clinical trials program. “Targeting ApoE, especially ApoE4, may be a promising therapeutic approach in reducing tau-mediated neurodegeneration,” the authors conclude.—Gabrielle Strobel
Q&A with David Holtzman
Q: You report a faster rate of progression in AD with ApoE4. Isn’t the current consensus view that E4 moves age of onset younger by as much as a decade, but does not accelerate the course of disease from there?
A: Yes, E4 moves age of onset by about five to 10 years on average per allele. This is almost for sure due to the effect of ApoE4 causing earlier onset of amyloid deposition. Much later, once people get cognitive impairment due to AD pathology, I am not aware that anyone has examined, as we did in this new work, how E4 affects rate of progression. We only evaluated people from ADNI and WashU who have very mild or mild dementia and have confirmed amyloid deposition by biomarkers. Without this confirmed biomarker, the finding we noted could be missed because individuals will be included who do not have AD.
Q: It seems the totality of your data supports reducing ApoE as a therapeutic target.
A: Yes, that’s what we think. The data from APP mice suggest one should lower ApoE as a “therapy.” The data from this new work suggests one should “lower” ApoE to decrease tau-mediated neurodegeneration. More research needs to be done now to determine whether you would still get the effect suggested in this new work if you lower ApoE in the brains of adults.
Q: Based on your new hypothesis, would targeting ApoE work later in the pathogenic process than targeting Aβ?
A: That’s what we are now trying to test.
Q: In other words, it could be easier to recruit for such trials, because you would not have to find participants, and assess drug effect, so many years prior to symptom onset.
A: That is exactly how I am thinking about it.
Q: The manifold interactions between ApoE and tau pathology—are they how “the disease becomes independent of Aβ”? We hear that phrase often these days.
A: We can’t be certain yet, but that is definitely a possibility.
Q: Activating microglia is sometimes proposed as a therapeutic approach, mainly to enhance their ability to phagocytose amyloid deposits. What does your data imply about this idea?
A: It may be that microglial activation is good to help abrogate amyloid deposition and its effects. However, for other causes of neurodegeneration that are not due to a protein accumulating extracellularly, such as tauopathies, synucleinopathies, etc., microglial activation may be quite deleterious. In other words, in AD, microglial activation may have stage-specific effects (good early, bad later). We are actively studying this question.
Q: Overall, how does this work change your view of ApoE in AD and other tauopathies?
A: I think it suggests we should now look at the role of ApoE in other tauopathies and other neurodegenerative diseases not on risk to get the disease, but whether ApoE isoforms/ApoE influences progression.
References
News Citations
Mutations Citations
Paper Citations
- Simonovitch S, Schmukler E, Bespalko A, Iram T, Frenkel D, Holtzman DM, Masliah E, Michaelson DM, Pinkas-Kramarski R. Impaired Autophagy in APOE4 Astrocytes. J Alzheimers Dis. 2016;51(3):915-27. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging Initiative, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017 Sep 28;549(7673):523-527. Epub 2017 Sep 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Pennsylvania
This is a very impressive study that revisits the link of ApoE4 to tau pathology, which was first examined by the Allen Roses group with Michel Goedert shortly after ApoE4 was identified as a risk factor for AD (Strittmatter et al., 1994). Oddly, there was little follow-up and the topic languished with regard to biological studies of the interactions of tau and ApoE, so this study certainly will reignite research that should have been followed up after the Strittmatter et al., 1994, paper.
References:
Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1994 Nov 8;91(23):11183-6. PubMed.
Hospital Clinic-IDIBAPS
Impressive study. However, it is still not clear what impact APOE genotype has in non-AD tauopathies in humans regarding clinical measures. In our series of 13 P301L MAPT patients (Borrego-Écija et al., 2017), the presence of the APOE E4 allele (four APOE4 carriers versus nine APOE4 non-carriers) did not influence the age of onset or duration of disease.
References:
Borrego-Écija S, Morgado J, Palencia-Madrid L, Grau-Rivera O, Reñé R, Hernández I, Almenar C, Balasa M, Antonell A, Molinuevo JL, Lladó A, Martínez de Pancorbo M, Gelpi E, Sánchez-Valle R. Frontotemporal Dementia Caused by the P301L Mutation in the MAPT Gene: Clinicopathological Features of 13 Cases from the Same Geographical Origin in Barcelona, Spain. Dement Geriatr Cogn Disord. 2017;44(3-4):213-221. Epub 2017 Sep 22 PubMed.
University of Arkansas for Medical Sciences
The link between APOE genotype and autophagy has recently received mechanistic reinforcement. We have demonstrated avid, specific binding of ApoE to DNA elements known as "CLEAR" sites, the cis elements bound by transcription factor EB (TFEB), a master regulator of autophagy-related genes (Parcon et al., 2017).
ApoE4 was found to bind more efficiently than ApoE3 through both empirical in vitro experiments and through computational modeling in silico analyses. The binding of CLEAR sites by ApoE4 appears to compete with binding by TFEB, thereby reducing the expression of autophagy genes. Evidence consistent with this was found in human brain samples, as well.
References:
Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ, Barger SW, Mrak RE, Griffin WS. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018 Feb;14(2):230-242. Epub 2017 Sep 22 PubMed.
Make a Comment
To make a comment you must login or register.