Tau Tracers Track First Emergence of Tangles in Familial Alzheimer’s
Quick Links
PET imaging transformed Alzheimer’s research when it enabled scientists to track the spread of Aβ pathology in living people. It revealed that amyloid plaques appear decades before symptoms of Alzheimer’s disease emerge. But what about neurofibrillary tangles? With the advent of tau tracers, scientists are now beginning to correlate the two major hallmarks of AD in real time. The results may clarify exactly how AD develops before a patient notices that anything is amiss. At the 10th annual Human Amyloid Imaging meeting, held January 13-15 in Miami Beach, Florida, preliminary data from two autosomal-dominant AD (ADAD) cohorts suggest that neurofibrillary tangles begin to accumulate much closer to the age of onset of symptoms than do amyloid plaques. “The data seem to suggest that in familial disease, tau tangles emerge long after Aβ but then rapidly spread,” said Reisa Sperling, Brigham and Women’s Hospital, Boston, a lead investigator on one of the studies. In short, when the tangles come, the real trouble starts.
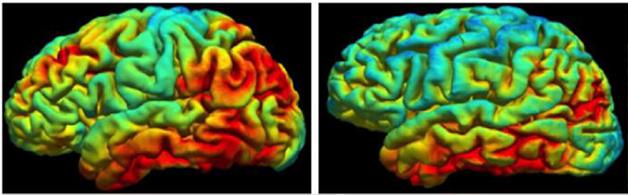
Familial vs. Sporadic. Tau pathology appears more widespread in autosomal-dominant (left) than sporadic AD. [Image courtesy of Tammie Benzinger.]
Scientists are curious to see how tangles develop in autosomal-dominant AD because patients are much younger at age of onset than those who develop sporadic AD, and likely have less age-related tau accumulation. Sperling collaborates on this study with Francisco Lopera and colleagues at the Universidad de Antioquia, Medellin, Colombia; Eric Reiman at Banner Health, Phoenix; and Yakeel Quiroz and Keith Johnson and colleagues at Massachusetts General Hospital. The scientists have scanned volunteers from a kindred in Colombia that lives with the presenilin1 E280A, aka Paisa, mutation.
Similarly, researchers led by Tammie Benzinger at Washington University, St. Louis, have been imaging tau deposits in families from the multinational DIAN observational study of familial AD. In her talk, Benzinger outlined early results for a group of 16 DIAN volunteers, both carriers and non-carriers of an FAD mutation. Because this sample size was so small, the DIAN study rules preclude divulging many details of these volunteers for fear of accidentally “unblinding" their genetic status and revealing private information. Benzinger presented just enough information to draw some tentative conclusions.
Benzinger compared the pattern of tau deposition in these DIAN volunteers with that seen in sporadic AD. Of the people so far scanned for tau, only two were classified as positive for tangles based on their uptake of the PET tracer AV1451. While the regional distribution of uptake in their brain was about the same as that seen among patients with sporadic AD, the amount seemed higher in the ADAD patients, Benzinger said. Both these DIAN participants were very mildly symptomatic, yet they had strikingly more AV1451 retention in the precuneus, parietal, and frontal lobes than sporadic patients with the same level of impairment, said Benzinger.
This might suggest more aggressive tau deposition in familial than in sporadic AD. However, that tau might be coming on quite late. The two tau-positive volunteers were within a few years of their expected age of onset. One cognitively normal volunteer with widespread Aβ deposition based on a PiB PET scan had a completely normal AV1451 scan. This person was 10 years younger than their predicted age of onset. By comparison, Aβ begins to deposit in the brain as much as 30 years before symptom onset.
For its part, the Colombian study suggests a similar timeline for tau. Analysis of the few scans done thus far hints that people with the Paisa mutation must be within about six years of age of onset before tau accumulates to detectable levels. In her poster, Quiroz described how she correlated PiB and AV1451 uptake in six asymptomatic PS1 mutation carriers and six age-matched non-carriers who last summer travelled from Colombia to Boston to be scanned. While the mutation carriers had higher cortical PiB uptake than same-age controls, no significant difference between the groups was evident on their AV1451 scans. Uptake of the tau ligand in the inferior temporal lobe and entorhinal cortex trended higher in mutation carriers than controls, as did an association between AV1451 uptake and amyloid burden. However, the latter trend was driven solely by one older mutation carrier, who was six years away from predicted age of onset in this cohort. The other five carriers were around age 30.
Sperling suggested that these patients’ relative youth may make them more resilient and that is why tangles do not emerge until fairly close to disease onset. But since FAD patients have more tau by the time mild symptoms start than do sporadic patients, their tau deposition must really ramp up quickly, she said. Quiroz agreed, and noted that age may play a role. “When thinking about sporadic AD, we have to consider that aging may confound the data,” she said. “People in their 70s already have tau pathology and have had it for many years, so some of the accumulation prior to onset is age-related.” That doesn’t happen with ADAD because the people are very young.
Both familial studies also suggest a lag between tau turning up in the cerebrospinal fluid and tangles forming in the brain. Previously WashU’s Ann Fagan and the DIAN team reported that CSF tau rises about 15 years before age of onset (Bateman et al., 2012). Last year Quiroz and colleagues reported the same in the Colombian kindred (see Jan 2015 news).
“We see the same mismatch in sporadic AD,” Benzinger told Alzforum. “I think we may ultimately decide that CSF and PET imaging are measuring two different aspects of tau pathophysiology.” She suggested that CSF tau may represent neuronal injury, which is why there are transient fluxes/increases in CSF tau in other instances of injury, such as trauma or stroke, while imaging captures the location of the abnormally folded neurofibrillary tangles. Quiroz has CSF samples from the 12 patients she scanned but has yet to run the analysis. She has applied for funding to run tests every 18 months on 30 mutation carriers and 30 controls.—Tom Fagan
References
Mutations Citations
News Citations
Paper Citations
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.