Location, Conformation, Decoration: Tau Biology Dazzles at AD/PD
Quick Links
The well of tau is in no danger of running dry, if findings presented at the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, are any gauge. If anything, it was bubbling over as scientists put forth a steady stream of data about the microtubule binding protein linked to neurodegeneration. There was the massive impact of tau pathology on the epigenome of neurons, as well as distinctive adornments peppering the protein in each tauopathy. Tau’s intimate, toxic affair with synaptic vesicles was on display, as was a mouse model in which Aβ-laden neuritic plaques sparked a wildfire spread of bona fide tau tangles throughout the brain. Scientists even awed the audience with high-resolution images of tau fibrils from human brain.
The breadth of findings should help researchers understand the impact of tau pathology on the brain, and how to target the protein with greater precision in therapies for neurodegenerative disease.
Does Tau Crack Open Chromatin?
Philip De Jager of Columbia University in New York described an impact of tau pathology on the epigenome, i.e., the collective series of DNA modifications that dictate which genes are expressed. Unlike the DNA sequence, which is static, the epigenome is subject to modification by its environment, and De Jager wanted to know how much sway Aβ and tau pathology held in that process. Working with David Bennett of Rush University Medical Center in Chicago, De Jager and colleagues extracted DNA from more than 600 postmortem dorsolateral prefrontal cortex samples from participants in the Religious Orders Study and the Memory and Aging Project. They mapped out more than 26,000 sites in the genome that were wrapped in acetylated histones—specifically H3K9Ac, a marker of open chromatin. The researchers then correlated a person’s acetylation pattern with the extent of his or her Aβ and tau pathology across five cortical regions. To De Jager’s surprise, tau’s impact on the epigenome topped that of Aβ’s by a full order of magnitude: Around 600 H3K9Ac marks correlated with Aβ, while nearly 6,000 correlated with tau. The intensity of these H3K9Ac peaks, which primarily landed in gene promoters or enhancers, correlated positively with transcription of associated genes.

Tau Pathology Opens DNA. A Manhattan plot of tau’s epigenetic influence across chromosome 1 reveals a pattern of “peaks of peaks,” in which tau influences large swaths of DNA. Each dot represents the size of tau’s effect on a single H3K9Ac peak. [Image courtesy of Philip De Jager.]
Was there a pattern to tau’s influence on the epigenome? Indeed, De Jager reported that while the epigenetic marks associated with Aβ were randomly scattered throughout the genome, those associated with tau clustered together in stretches containing hundreds of genes. The tau-associated H3K9Ac marks also coincided with binding sites for CTCF, a protein that regulates chromatin structure. The findings suggested that tau pathology somehow triggered large-scale alterations in chromatin structure—opening access to and enabling transcription of large swaths of the genome.
De Jager confirmed these findings in mice overexpressing tau and by forcing tau overexpression in human iPSC-derived neurons. On the flip side, the researchers blocked tau’s epigenetic influence by treating tau overexpressing neurons with the Hsp90 inhibitors alvespimycin or geldanamycin—two compounds predicted to reduce the effects of tau pathology. The impact of tau’s epigenomic effect on neurodegeneration is unclear, but De Jager proposed that elevated expression of so many genes could perhaps tie up the transcription machinery, leading to broad malfunction across the cell.
Even as tau pathology affects gene expression, the tau gene itself (aka MAPT) is not immune to regulation, either. According to Roberto Simone of University College London, a sequence in MAPT’s own 5‛ untranslated region keeps levels of the protein in check. Sequencing RNA from human brain samples and iPSC-derived neurons, Simone uncovered an antisense long non-coding RNA (lncRNA) called MAPT-AS1 that inhibited tau translation. LncRNAs, which are typically greater than 200 nucleotides in length, are emerging players in gene regulation, and their importance in neurodegenerative disease is only just beginning to surface (reviewed in Luo and Chen, 2016). Speaking at AD/PD, Simone suggested to the audience that MAPT-AS1 adheres to the tau mRNA’s internal ribosome entry site, where the lncRNA fends off approaching ribosomes via a mammalian-wide interspersed repeat (MIR) element. Silencing MAPT-AS1 boosted tau expression, while stable expression of the lncRNA in neuroblastoma cell lines repressed it, Simone reported. While this finding raises the idea that tau proteostasis could one day be influenced via lncRNAs for therapeutic purposes, basic research on these regulatory RNAs is in its early stages.
Tau: Form and Function
After tau translation is said and done, the resulting protein is subject to a slew of post-translational modifications, some of which contribute to its neurodegenerative power. While specific antibodies help researchers measure some of these adornments, such as phosphorylation of certain residues, these approaches do not draw a comprehensive map of modifications along the entire protein. At AD/PD, Judith Steen of Boston Children’s Hospital presented findings rendered from a mass spectroscopy-based technique called FLEXITau, which maps both the nature and quantity of tau modifications. Using an isotope-labeled version of tau as a standard, the method previously enabled Steen to map tau modifications in the Alzheimer’s brain (see Mair et al., 2016).
In Vienna, Steen described follow-up work using FLEXITau to compare modifications across different tauopathies. Analyzing 129 postmortem brain samples from people with AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), or Pick’s disease (PiD), Steen found that tau in each disease had its own distinctive brand of phosphorylation, acetylation, ubiquitination, and fragmentation patterns. Based on these signatures, Steen developed an algorithm that correctly classified disease type in 90 percent of AD, CBD, PiD, and control samples, and around 80 percent of PSP samples. Steen is currently investigating whether FLEXITau done on CSF or plasma tau might work as a diagnostic tool. More importantly, she told Alzforum that understanding disease-related modifications should help researchers develop antibodies that specifically target pathological forms of tau in each disease.
Besides donning decorations, the tau protein also strikes a dizzying array of poses.
While neurodegeneration often accompanies its most infamous form—neurofibrillary tangles—researchers have long noted that synaptic defects, neuroinflammation, and even neuronal loss can occur in cells free of tangles, and before tangles are widespread (see Gomez-Isla et al., 1997; Feb 2007 news). Paralleling the Aβ story, researchers now think that soluble tau oligomers might be the silent culprit (see Berger et al., 2007; Brunden et al., 2008; Meraz-Rios et al., 2010; and Nov 2010 conference news).
In support of this idea, Eva and Eckhard Mandelkow of the German Center for Neurodegenerative Diseases in Bonn presented findings describing the structure and toxicity of small tau oligomers. They used bacterial cells as factories to pump out recombinant tauRDΔK, an aggregation-prone version of the full-length neurotoxic protein containing four repeat domains. They then tinkered with incubation buffers to coax the protein to oligomerize, and found that the resulting oligomers took on a globular shape, consisting primarily of dimers, trimers, and tetramers. When they purified these oligomers and added them to hippocampal neurons, the little tau blobs ramped up production of reactive oxygen species (ROS), boosted intracellular calcium, and reduced the density of dendritic spines in the neurons. However, the oligomers stopped short of killing the neurons. The Mandelkows proposed that soluble tau oligomers contribute to synaptic defects and inflammation as first steps on the road to neurodegeneration. In addition to globular oligomers, the Mandelkows observed more ordered filamentous tau aggregates in their protein preps.
Fibrillar forms of tau, including the paired helical filaments that dominate neurofibrillary tangles and the straight protofibrils that precede them, were the object of the first atomic-level structures of tau ever reported. At AD/PD, Anthony Fitzpatrick of the University of Cambridge, U.K., had a spellbound audience when he described 3.4 A resolution, cryo-EM structures of tau aggregates isolated from patient brain. He showed protofibrils, which were marked by β-solenoid structures previously described for prions. Alzforum will discuss these structures in detail when they are formally published. In a nutshell, Fitzpatrick in Vienna described a fibril structure held together by C-shaped protofilaments made up of tau’s R3 and R4 repeat domains. Tau’s first two repeats, as well as the protein’s N-terminus, formed a “fuzzy coat” around this R3/R4 core. Intriguingly, the structures included a putative binding site for the PET tracer AV1451/flortaucipir, and they will likely help inform structure-based drug design. These structures mark a new era in tau science, Eckhard Mandelkow told Alzforum.
Selina Wray of University College, London, called Fitzpatrick’s talk the highlight of the conference. She was particularly excited that the work may help explain how different structural polymorphisms of tau relate to the diversity of tau strains and pathologies.
Tau: Scene of the Crime
Tau’s effects on dendrites, and the postsynapses that sit on the dendrites, have been well-studied, partly because normal tau is scarce in those regions of the neuron and tau’s presence there struck researchers as a pathological target. However, some researchers, including the Mandelkows, have discovered that tau can also misbehave on its home turf. They previously reported that in model mice, tau aggregates stray from tau’s main site in axons into axon terminals, where they trigger synaptic dysfunction and loss from the presynaptic side (see Apr 2015 news).
In Vienna, Joseph McInnes of VIB KU Leuven, Belgium put forward a mechanism for this finding. Working with Patrik Verstreken and Bart De Strooper, McInnes examined the neuromuscular junctions of flies that express human wild-type or mutant tau, including P301L, V337M, and R406W. In flies with a mutant gene, tau aggregates accumulated at presynapses. The presence of tau there correlated with impaired vesicle fusion and neurotransmitter release. To find out why, McInnes turned to in vitro assays of purified tau and vesicles, finding that tau’s N-terminal domain bound the vesicles. In the fly synapses, tau aggregates in effect tied up vesicles, clustering them and preventing their release. Supporting this, a mutant tau transgene that lacked the N-terminal domain still localized to presynapses but did not harm function.

Vesicle Interloper?
Nanogold-labeled recombinant human Tau clings to synaptic vesicles (SV) isolated from the rat brain. [Image courtesy of Joseph McInnes.]
McInnes verified the finding in cultures of rat primary hippocampal and human neurons. When he added a peptide that competes with tau to bind vesicles, he prevented synaptic toxicity. He also detected high levels of phosphotau in synaptic vesicles isolated from postmortem AD hippocampal samples, suggesting the same mechanism could be at work in people. McInnes told Alzforum that he has zeroed in on a vesicular protein that latches onto tau in the presynapses. He aims to disentangle the relative contributions of pre- and postsynaptic tau to synaptoxicity by knocking it down.
Intrigued by McInnes’ finding, the Mandelkows pointed out that, similar to the role of tau in axons, the N-terminus of tau is relatively understudied compared to its C-terminus, which contains the repeat domains that bind microtubules and facilitate aggregation. “One known feature [of the N-terminus] is that it mediates the binding of tau to motor proteins and the axonal cytoskeleton, e.g. through dynactin, which would make sense in the context of axonal trafficking of synaptic vesicles,” they wrote to Alzforum (see Magnani et al., 2007).
A Better Mouse of Traveling Tau?
One hallmark of human tau neuropathology is its staged spread throughout the brain; alas, this has been difficult to recapitulate fully in mice because many mouse models of AD develop no neurofibrillary tangles, let alone tangles that propagate. This is true of models that express human mutant APP in a mouse tau background, such as APPPS1. More puzzling still, it even holds true in mice that express wild-type human tau in the presence of amyloid, or when human neurons are transplanted into AD mouse brain (see Dec 2016 conference news; Feb 2017 news). Some researchers have suggested that this may be because tangles require seeding to form.
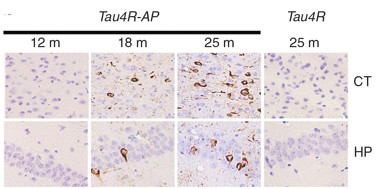
Toxic Templating.
In APPPS1 mice expressing a fragment of four-repeat tau (Tau4R-AP), endogenous mouse tau by 18 months transformed into tangles that exacerbated at 25 months, both in cortex (CT) and hippocampus (HP). Single transgenic tau4R mice (right panel) developed no tangles. [Image courtesy of Li et al., Nature Communications 2016.]
At AD/PD, Philip Wong of Johns Hopkins University, Baltimore, presented evidence to back this idea. He generated mice that express an inducible fragment from the four-repeat isoform of human tau, which can be turned off by dietary tetracycline. The animals remained healthy, with no tau pathology, but when Wong crossed them to APPPS1 mice, the offspring developed neurofibrillary tangles throughout their hippocampi and cortices (see Li et al., 2016). Wong determined that these tangles, and their spread, started after Aβ-laden neuritic plaques had formed. Wong saw dramatic neuronal loss in the hippocampus and cortex at 15 months, along with extensive microgliosis and astrogliosis. He suggested that four-repeat tau may act as a template for misfolding wild-type tau, but that this happens only in the presence of neuritic amyloid plaques. Wong plans to switch off expression of the seed before or after wild-type mouse tau starts aggregating, to test if tau pathology can continue unabated once sparked. This mouse model could be useful for testing therapeutic strategies, he added.
Why did previous models using full-length human tau not convert tau into tangles and show spread of this process? Wong told Alzforum that perhaps tau fragmentation is necessary to create a proteopathic seed. This fragmentation may unfold over decades in humans, and mice expressing full-length human tau might not live long enough for it to happen, he said.—Jessica Shugart and Madolyn Bowman Rogers
References
News Citations
- Tau Toxicity—Tangle-free But Tied to Inflammation
- San Diego: Tau Oligomer Antibodies Relieve Motor Deficits in Mice
- Not All About Dendrites: Presynaptic Tau Harms Plasticity, Too
- Next-Generation Mouse Models: Tau Knock-ins and Human Chimeras
- Chimeric AD Model Shows Human Neurons Are Uniquely Vulnerable to Aβ
Mutations Citations
Research Models Citations
Paper Citations
- Luo Q, Chen Y. Long noncoding RNAs and Alzheimer's disease. Clin Interv Aging. 2016;11:867-72. Epub 2016 Jun 29 PubMed.
- Mair W, Muntel J, Tepper K, Tang S, Biernat J, Seeley WW, Kosik KS, Mandelkow E, Steen H, Steen JA. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal Chem. 2016 Apr 5;88(7):3704-14. Epub 2016 Mar 7 PubMed.
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997 Jan;41(1):17-24. PubMed.
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007 Apr 4;27(14):3650-62. PubMed.
- Brunden KR, Trojanowski JQ, Lee VM. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008 Aug;14(4):393-9. PubMed.
- Meraz-Ríos MA, Lira-De León KI, Campos-Peña V, De Anda-Hernández MA, Mena-López R. Tau oligomers and aggregation in Alzheimer's disease. J Neurochem. 2010 Mar;112(6):1353-67. PubMed.
- Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. 2007 Oct 31;26(21):4546-54. PubMed.
- Li T, Braunstein KE, Zhang J, Lau A, Sibener L, Deeble C, Wong PC. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer's disease mouse model. Nat Commun. 2016 Jul 4;7:12082. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.