Bad Origami: Misfolded SOD1 in Living Color
Quick Links
When proteins fail to curl up in the right conformation, molecular chaperones sort them out, or the misfits are tagged for destruction. But if chaperones and the proteasome are unable to control a misfolded protein, disaster results. Misfolded, aggregated proteins are a common feature of neurodegenerative diseases and a major focus of research. This month, two papers from the laboratory of Arthur Horwich, at the Yale University School of Medicine in New Haven, Connecticut, explore how misfolded copper-zinc superoxide dismutase 1 (SOD1) wreaks havoc by building up in new worm and mouse models of amyotrophic lateral sclerosis (ALS). Horwich’s lab is known for studying chaperones; this is the group’s first foray into ALS research.
By tagging the protein with yellow fluorescent protein (YFP), the scientists were able to track its accumulation in neurons as symptoms progressed. “It’s just solid fluorescence,” said George Farr of the affected neurons in the mouse model. Farr, also of Yale School of Medicine, is joint first author on the mouse paper, published online in PNAS January 26. The aggregates eventually outgrow the cells they came from and join together, he said, in a microscopic scene of devastation reminiscent of the ’80s horror flick The Blob. The new models will allow scientists to identify genes and proteins that interact with SOD1 aggregates, said Jiou Wang, first author on the PNAS paper and the worm paper, published in PLoS Genetics on January 23.
Mutant SOD1 is responsible for disease in approximately 2 percent of people with ALS. Scientists have identified more than 100 SOD1 mutations in these cases of inherited ALS. They also know that the mutations cause a toxic gain of function in the enzyme, but exactly how neurons then die remains unclear (for review, see Furukawa and O’Halloran, 2006). People with ALS caused by SOD1 mutation, and also some people with sporadic cases, show SOD1 aggregates in the spinal cord (Shibata et al., 1996).
Great globs of misfolded proteins are a common feature in neurodegenerative disease—Alzheimer’s has Aβ plaques and tau tangles, Parkinson’s has Lewy bodies, Huntington’s has ubiquitinated huntingtin inclusions, and many of these diseases have TDP-43 deposits. Horwich wondered why these damaged goods cause so much trouble, since the cell has chaperones and proteasomes to mend or destroy the wayward peptides. “It’s disturbing to me that SOD1 could actually misfold…despite the presence of molecular chaperones,” Horwich said. In cell culture, studies have shown that the human SOD1-G85R mutant forms aggregates and associates with the chaperone Hsp70 (Matsumoto et al., 2005). To examine SOD1 misfolding in vivo, the Yale scientists developed YFP-tagged constructs of the same mutant. G85R is a seriously misfolded, inactive protein, which fails to form a disulfide bond and dimerize, as wild-type SOD1 does (Jonsson et al., 2006).
Sluggish Worms
SOD1 mice have led to many discoveries, but even so, “the field really needs a simpler and faster model organism,” Wang said. Mice take weeks to gestate and months to develop disease, making the kind of large-scale genetic screens that Wang wants to do nearly impossible. Of the few existing invertebrate models for mSOD1-based ALS (e.g., Watson et al., 2008; Witan et al., 2008; Oeda et al., 2001), none quite matched Wang’s needs.
In the PLoS Genetics paper, Wang and colleagues describe a transgenic Caenorhabditis elegans model carrying human SOD1-G85R-YFP under the control of a general neural promoter. Compared to worms carrying wild-type SOD1, the mutants show a severe movement phenotype. “It’s like they’re all post-Thanksgiving dinner; they’re just lazing around the plate,” Farr said. In contrast, wild-type animals wriggle energetically across the medium.
In the neurons of wild-type animals, SOD1-YFP showed a diffuse cytosolic distribution, whereas in the mutants, the fluorescent proteins glommed together. Ventral nerve cord cells were shriveled, with fewer organelles and processes. The mutant worms also had fewer synapses and synaptic vesicles, suggesting to the scientists that they have a presynaptic defect. Some of the transgenic worms lived a nearly normal lifespan (about three weeks), however, unlike mice or people with mutant SOD1. In mice, the loss of motor function ultimately prevents the animals from eating and drinking; at this stage, scientists usually sacrifice the animals. The problem may not be so severe in the worms, Wang suggested, because they only need to move a bit to be able to slurp up bacteria from the surrounding media.
Wang and colleagues then used the worms to fish for genes that affect SOD1 accumulation. An RNA interference screen netted genes that, when silenced, cause SOD1 aggregation to go down or up. In the “down” category, the scientists identified a ubiquitin protease. In the “up” category, they pulled out players in TGF-β signaling, DNA replication and repair, and RNA transcription. Several more hits corresponded to molecular chaperones, confirming that chaperones help reduce protein aggregation. The scientists plan to validate candidate genes for involvement in SOD1 aggregation, and then mutate those candidates in mouse models for further study.
Chock Full of SOD
The mouse model described in the PNAS paper is the first animal to express fluorescent SOD1, Horwich said. Transgenic mice homozygous for the G85R mutant developed motor dysfunction at approximately nine months of age, whereas wild-type animals and heterozygotes appeared healthy. Heterozygotes probably did not express enough of the transgene to cause symptoms, Farr said.
Mice with wild-type SOD1-YFP showed low levels of fluorescence throughout their lifespan of more than two years. The signal was stronger in SOD1-G85R-YFP mice, and intensified as the animals aged. At one and six months of age, the mutant animals exhibited diffuse staining across neurons, particularly in the anterior horn. But at nine months, once the animals began to sicken, the researchers observed intense, punctate staining in many neural cell bodies.
The scientists then used biochemical fractionation to follow the appearance of insoluble SOD1 in the spinal cord. SOD1 was uniformly soluble in wild-type animals, and in one-month-old G85R mice. But by six months, one-quarter of SOD1-YFP in the mutant animals was insoluble. By nine months, half of the fusion protein was insoluble.
In the soluble protein fraction, SOD1-G85R-YFP from one-month-old mutants appeared at approximately 45 kDa on a Western blot, the expected size of a monomer. But in older animals, larger species appeared. These likely correspond to oligomers of misfolded SOD1, the authors said. However, the SOD1 complexes did not grow in a stepwise fashion, with steady increases of 45 kDa each. That suggested to Farr and colleagues that other proteins were associating with the SOD1 oligomers, resulting in aggregates of varying sizes.
The scientists turned to proteomics to identify the hangers-on and found that soluble SOD1-G85R-YFP linked to several other proteins. Most abundant among the associates, at all ages, was the chaperone Hsc70. In older animals, three chaperones from the Hsp110 family joined in, showing that chaperones do bind the misfolded SOD. In younger animals, mutant SOD1 poses no problem, because the cell is able to ubiquitinate and degrade the mutant SOD1 within a few days. “It’s getting chewed on pretty well,” Horwich said. “It seems like the systems are doing the best they can to get rid of it.” The authors speculate that as the animals age, however, mutant SOD1 may build up and overwhelm cellular defenses. “It gets to the point where the [oligomers] are so large that they just fall out of solution altogether, and then the cell body of the neuron is in really big trouble,” Farr said.
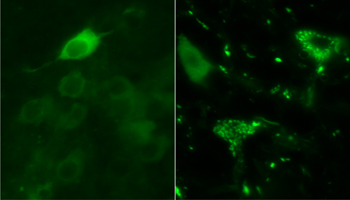
Great Globs of SOD
YFP-tagged mutant SOD1 is cytoplasmic in one-month-old animals (left), while it forms forms bright, punctate aggregates in motor neurons of 10-month-old transgenic mice. Image credit: George Farr
This research confirms that molecular chaperones act as the first line of defense against growing globs of mutant SOD1, Horwich said. He suspects that an extra dose of chaperone might hold off the aggregates for longer, and plans to test the hypothesis in model systems.
The current studies do not address how, and if, aggregated SOD1 is deadly to cells. “I’d dearly like to know what the toxicity is,” Horwich said. At least with his new animal models, he has further tools to pursue that question.
The strength of Horwich’s work lies in the combination of imaging, biochemistry, and proteomics, said Rick Morimoto of Northwestern University in Evanston, Illinois, who was not an author of the current papers. This combination allowed the Yale group to dissect SOD1 accumulation in detail. Morimoto further suggested that chaperones losing control of concentrated misfolded proteins could be a common feature among neurodegenerative disorders. “If you have an accumulation of damaged protein, and then you have decreased capacity of the chaperone networks happening at the same time, you can well imagine that is a catastrophic recipe,” he said.—Amber Dance
References
Paper Citations
- Furukawa Y, O'Halloran TV. Posttranslational modifications in Cu,Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006 May-Jun;8(5-6):847-67. PubMed.
- Shibata N, Asayama K, Hirano A, Kobayashi M. Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev Neurosci. 1996;18(5-6):492-8. PubMed.
- Matsumoto G, Stojanovic A, Holmberg CI, Kim S, Morimoto RI. Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J Cell Biol. 2005 Oct 10;171(1):75-85. PubMed.
- Jonsson PA, Graffmo KS, Andersen PM, Brännström T, Lindberg M, Oliveberg M, Marklund SL. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006 Feb;129(Pt 2):451-64. PubMed.
- Oeda T, Shimohama S, Kitagawa N, Kohno R, Imura T, Shibasaki H, Ishii N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum Mol Genet. 2001 Sep 15;10(19):2013-23. PubMed.
Further Reading
Papers
- Watanabe Y, Morita E, Fukada Y, Doi K, Yasui K, Kitayama M, Nakano T, Nakashima K. Adherent monomer-misfolded SOD1. PLoS One. 2008;3(10):e3497. PubMed.
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006 Mar 10;311(5766):1471-4. PubMed.
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998 Sep 18;281(5384):1851-4. PubMed.
- Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002 Oct;4(10):826-31. PubMed.
Primary Papers
- Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, Perrin S, Feany MB, Horwich AL. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009 Feb 3;106(5):1392-7. PubMed.
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009 Jan;5(1):e1000350. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.