Mutations
MAPT L284L
Quick Links
Overview
Pathogenicity: Frontotemporal Dementia : Pathogenic
Clinical
Phenotype: Frontotemporal Dementia
Reference Assembly: GRCh37/hg19
Position: Chr17:44087705 T>C
dbSNP ID: rs63751423
Coding/Non-Coding: Coding
DNA
Change: Substitution
Expected RNA
Consequence: Splicing Alteration
Expected Protein
Consequence: Isoform Shift; Silent
Codon
Change: CTT to CTC
Reference
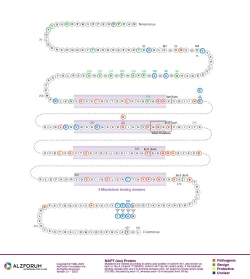
Isoform: Tau Isoform Tau-F (441 aa)
Genomic
Region: Exon 10
Findings
This silent mutation was identified in a family known as LKL, whose members were affected by a form of early onset dementia. Their disease was identified as a variant of frontotemporal dementia with difficulties in word-finding and visual-spatial abilities, behavioral changes, and a decline in executive functioning. The reported pedigree shows six affected individuals over three generations. The mean age at onset was 51.8 ± 4.8 years. Symptoms commonly included forgetfulness, confusion, and lack of insight. The mutation was shown to segregate with disease in this family; it was present in two affected individuals and absent in the mother of one of the affected individuals who was apparently affected by sporadic AD (she developed AD at age 75 and had an APOE genotype of ε4/ε4). The mutation was not present in 96 controls. No additional mutations were detected in MAPT exons, or in PSEN1, PSEN2, or APP in the three LKL family members screened (D'Souza et al., 1999).
Neuropathology
Autopsy results from one affected mutation carrier showed moderate bilateral frontal and right temporal atrophy and a variety of tau aggregates in both neurons and glia. Specifically, neurofibrillary tangles were observed in the neocortex, amygdala, and parahippocampus. Although neurofibrillary tangles were relatively sparse in the hippocampus, a large numbers of argyrophilic grains and neuropil threads were observed there. Tau-positive glia were observed in the white matter. Aβ immunostaining showed numerous diffuse plaques in the neocortex as well as a smaller number of neuritic plaques with dense cores. Overall, the amyloid pathology and other neuropathological findings met CERAD criteria for AD, and the authors note that coincident AD was a possibility (D'Souza et al., 1999).
Biological Effect
The silent L284L mutation is associated with an increase in transcripts containing exon 10 and a decrease in transcripts lacking exon 10. The mutation is thought to destroy an exon-splicing silencing element (D'Souza et al., 1999).
Last Updated: 16 Feb 2023
References
Paper Citations
- D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999 May 11;96(10):5598-603. PubMed.
Further Reading
Papers
- Poorkaj P, Grossman M, Steinbart E, Payami H, Sadovnick A, Nochlin D, Tabira T, Trojanowski JQ, Borson S, Galasko D, Reich S, Quinn B, Schellenberg G, Bird TD. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch Neurol. 2001 Mar;58(3):383-7. PubMed.
Learn More
Protein Diagram
Primary Papers
- D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999 May 11;96(10):5598-603. PubMed.
Other mutations at this position
Alzpedia
Disclaimer: Alzforum does not provide medical advice. The Content is for informational, educational, research and reference purposes only and is not intended to substitute for professional medical advice, diagnosis or treatment. Always seek advice from a qualified physician or health care professional about any medical concern, and do not disregard professional medical advice because of anything you may read on Alzforum.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.